
Introduction
Scientific studies have found that different brain areas show altered activity in people with major depressive disorder (MDD), and this has encouraged advocates of various theories that seek to identify a biochemical origin of the disease, as opposed to theories that emphasize psychological or situational causes.

Factors spanning these causative groups include nutritional deficiencies in magnesium, vitamin D, and tryptophan with situational origin but biological impact. Several theories concerning the biologically based cause of depression have been suggested over the years, including theories revolving around monoamine neurotransmitters, neuroplasticity, neurogenesis, inflammation and the circadian rhythm. Physical illnesses, including hypothyroidism and mitochondrial disease, can also trigger depressive symptoms.
Neural circuits implicated in depression include those involved in the generation and regulation of emotion, as well as in reward. Abnormalities are commonly found in the lateral prefrontal cortex whose putative function is generally considered to involve regulation of emotion. Regions involved in the generation of emotion and reward such as the amygdala, anterior cingulate cortex (ACC), orbitofrontal cortex (OFC), and striatum are frequently implicated as well. These regions are innervated by a monoaminergic nuclei, and tentative evidence suggests a potential role for abnormal monoaminergic activity.
Genetic Factors
Difficulty of Gene Studies
Historically, candidate gene studies have been a major focus of study. However, as the number of genes reduces the likelihood of choosing a correct candidate gene, Type I errors (false positives) are highly likely. Candidate genes studies frequently possess a number of flaws, including frequent genotyping errors and being statistically underpowered. These effects are compounded by the usual assessment of genes without regard for gene-gene interactions. These limitations are reflected in the fact that no candidate gene has reached genome-wide significance.
Gene Candidates
5-HTTLPR
The 5-HTTLPR, or serotonin transporter promoter gene’s short allele, has been associated with increased risk of depression; since the 1990s, however, results have been inconsistent. Other genes that have been linked to a gene-environment interaction include CRHR1, FKBP5 and BDNF, the first two of which are related to the stress reaction of the HPA axis, and the latter of which is involved in neurogenesis. Candidate gene analysis of 5-HTTLPR on depression was inconclusive on its effect, either alone or in combination with life stress.
A 2003 study proposed that a gene-environment interaction (GxE) may explain why life stress is a predictor for depressive episodes in some individuals, but not in others, depending on an allelic variation of the serotonin-transporter-linked promoter region (5-HTTLPR). This hypothesis was widely-discussed in both the scientific literature and popular media, where it was dubbed the “Orchid gene”, but has conclusively failed to replicate in much larger samples, and the observed effect sizes in earlier work are not consistent with the observed polygenicity of depression.
BDNF
BDNF polymorphisms have also been hypothesized to have a genetic influence, but early findings and research failed to replicate in larger samples, and the effect sizes found by earlier estimates are inconsistent with the observed polygenicity of depression.
SIRT1 and LHPP
A 2015 GWAS study in Han Chinese women positively identified two variants in intronic regions near SIRT1 and LHPP with a genome-wide significant association.
Norepinephrine Transporter Polymorphisms
Attempts to find a correlation between norepinephrine transporter polymorphisms and depression have yielded negative results.
One review identified multiple frequently studied candidate genes. The genes encoding for the 5-HTT and 5-HT2A receptor were inconsistently associated with depression and treatment response. Mixed results were found for brain-derived neurotrophic factor (BDNF) Val66Met polymorphisms. Polymorphisms in the tryptophan hydroxylase gene was found to be tentatively associated with suicidal behaviour. A meta analysis of 182 case controlled genetic studies published in 2008 found Apolipoprotein E verepsilon 2 to be protective, and GNB3 825T, MTHFR 677T, SLC6A4 44bp insertion or deletions, and SLC6A3 40 bpVNTR 9/10 genotype to confer risk.
Circadian Rhythm
Depression may be related to abnormalities in the circadian rhythm, or biological clock.
A well synchronised circadian rhythm is critical for maintaining optimal health. Adverse changes and alterations in the circadian rhythm have been associated various neurological disorders and mood disorders including depression.

Sleep
Sleep disturbance is the most prominent symptom in depressive patients. Studies about sleep electroencephalograms have shown characteristic changes in depression such as reductions in non-rapid eye movement sleep production, disruptions of sleep continuity and disinhibition of rapid eye movement (REM) sleep. Rapid eye movement (REM) sleep – the stage in which dreaming occurs – may be quick to arrive and intense in depressed people. REM sleep depends on decreased serotonin levels in the brain stem, and is impaired by compounds, such as antidepressants, that increase serotonergic tone in brain stem structures. Overall, the serotonergic system is least active during sleep and most active during wakefulness. Prolonged wakefulness due to sleep deprivation activates serotonergic neurons, leading to processes similar to the therapeutic effect of antidepressants, such as the selective serotonin reuptake inhibitors (SSRIs). Depressed individuals can exhibit a significant lift in mood after a night of sleep deprivation. SSRIs may directly depend on the increase of central serotonergic neurotransmission for their therapeutic effect, the same system that impacts cycles of sleep and wakefulness.
Light Therapy
Research on the effects of light therapy on seasonal affective disorder suggests that light deprivation is related to decreased activity in the serotonergic system and to abnormalities in the sleep cycle, particularly insomnia. Exposure to light also targets the serotonergic system, providing more support for the important role this system may play in depression. Sleep deprivation and light therapy both target the same brain neurotransmitter system and brain areas as antidepressant drugs, and are now used clinically to treat depression. Light therapy, sleep deprivation and sleep time displacement (sleep phase advance therapy) are being used in combination quickly to interrupt a deep depression in people who are hospitalised for MDD.
Increased and decreased sleep length appears to be a risk factor for depression. People with MDD sometimes show diurnal and seasonal variation of symptom severity, even in non-seasonal depression. Diurnal mood improvement was associated with activity of dorsal neural networks. Increased mean core temperature was also observed. One hypothesis proposed that depression was a result of a phase shift.
Daytime light exposure correlates with decreased serotonin transporter activity, which may underlie the seasonality of some depression.
Monoamines
Monoamines are neurotransmitters that include serotonin, dopamine, norepinephrine, and epinephrine.
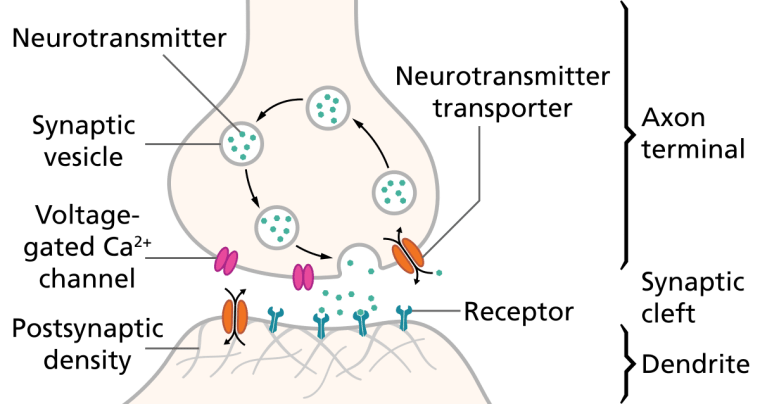
Monoamine Hypothesis of Depression
Many antidepressant drugs acutely increase synaptic levels of the monoamine neurotransmitter, serotonin, but they may also enhance the levels of norepinephrine and dopamine. The observation of this efficacy led to the monoamine hypothesis of depression, which postulates that the deficit of certain neurotransmitters is responsible for depression, and even that certain neurotransmitters are linked to specific symptoms. Normal serotonin levels have been linked to mood and behaviour regulation, sleep, and digestion; norepinephrine to the fight-or-flight response; and dopamine to movement, pleasure, and motivation. Some have also proposed the relationship between monoamines and phenotypes such as serotonin in sleep and suicide, norepinephrine in dysphoria, fatigue, apathy, cognitive dysfunction, and dopamine in loss of motivation and psychomotor symptoms.[31] The main limitation for the monoamine hypothesis of depression is the therapeutic lag between initiation of antidepressant treatment and perceived improvement of symptoms. One explanation for this therapeutic lag is that the initial increase in synaptic serotonin is only temporary, as firing of serotonergic neurons in the dorsal raphe adapt via the activity of 5-HT1A autoreceptors. The therapeutic effect of antidepressants is thought to arise from autoreceptor desensitization over a period of time, eventually elevating firing of serotonergic neurons.
Serotonin
Initial studies of serotonin in depression examined peripheral measures such as the serotonin metabolite 5-Hydroxyindoleacetic acid (5-HIAA) and platelet binding. The results were generally inconsistent, and may not generalise to the central nervous system. However evidence from receptor binding studies and pharmacological challenges provide some evidence for dysfunction of serotonin neurotransmission in depression. Serotonin may indirectly influence mood by altering emotional processing biases that are seen at both the cognitive/behavioural and neural level. Pharmacologically reducing serotonin synthesis, and pharmacologically enhancing synaptic serotonin can produce and attenuate negative affective biases, respectively. These emotional processing biases may explain the therapeutic gap.
Dopamine
While various abnormalities have been observed in dopaminergic systems, results have been inconsistent. People with MDD have an increased reward response to dextroamphetamine compared to controls, and it has been suggested that this results from hypersensitivity of dopaminergic pathways due to natural hypoactivity. While polymorphisms of the D4 and D3 receptor have been implicated in depression, associations have not been consistently replicated. Similar inconsistency has been found in post-mortem studies, but various dopamine receptor agonists show promise in treating MDD. There is some evidence that there is decreased nigrostriatal pathway activity in people with melancholic depression (psychomotor retardation). Further supporting the role of dopamine in depression is the consistent finding of decreased cerebrospinal fluid and jugular metabolites of dopamine, as well as post mortem findings of altered Dopamine receptor D3 and dopamine transporter expression. Studies in rodents have supported a potential mechanism involving stress-induced dysfunction of dopaminergic systems.
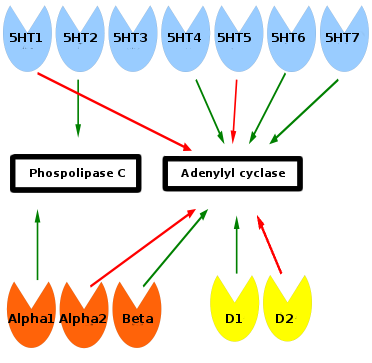
Catecholamines
A number of lines of evidence indicative of decreased adrenergic activity in depression have been reported. Findings include the decreased activity of tyrosine hydroxylase, decreased size of the locus coeruleus, increased alpha 2 adrenergic receptor density, and decreased alpha 1 adrenergic receptor density. Furthermore, norepinephrine transporter knockout in mice models increases their tolerance to stress, implicating norepinephrine in depression.
One method used to study the role of monoamines is monoamine depletion. Depletion of tryptophan (the precursor of serotonin), tyrosine and phenylalanine (precursors to dopamine) does result in decreased mood in those with a predisposition to depression, but not in persons lacking the predisposition. On the other hand, inhibition of dopamine and norepinephrine synthesis with alpha-methyl-para-tyrosine does not consistently result in decreased mood.
Monoamine Oxidase
An offshoot of the monoamine hypothesis suggests that monoamine oxidase A (MAO-A), an enzyme which metabolises monoamines, may be overly active in depressed people. This would, in turn, cause the lowered levels of monoamines. This hypothesis received support from a PET study, which found significantly elevated activity of MAO-A in the brain of some depressed people. In genetic studies, the alterations of MAO-A-related genes have not been consistently associated with depression. Contrary to the assumptions of the monoamine hypothesis, lowered but not heightened activity of MAO-A was associated with depressive symptoms in adolescents. This association was observed only in maltreated youth, indicating that both biological (MAO genes) and psychological (maltreatment) factors are important in the development of depressive disorders. In addition, some evidence indicates that disrupted information processing within neural networks, rather than changes in chemical balance, might underlie depression.
Limitations
Since the 1990s, research has uncovered multiple limitations of the monoamine hypothesis, and its inadequacy has been criticised within the psychiatric community. For one thing, serotonin system dysfunction cannot be the sole cause of depression. Not all patients treated with antidepressants show improvements despite the usually rapid increase in synaptic serotonin. If significant mood improvements do occur, this is often not for at least two to four weeks. One possible explanation for this lag is that the neurotransmitter activity enhancement is the result of auto receptor desensitization, which can take weeks. Intensive investigation has failed to find convincing evidence of a primary dysfunction of a specific monoamine system in people with MDD. The antidepressants that do not act through the monoamine system, such as tianeptine and opipramol, have been known for a long time. There have also been inconsistent findings with regard to levels of serum 5-HIAA, a metabolite of serotonin. Experiments with pharmacological agents that cause depletion of monoamines have shown that this depletion does not cause depression in healthy people. Another problem that presents is that drugs that deplete monoamines may actually have antidepressant properties. Further, some have argued that depression may be marked by a hyperserotonergic state. Already limited, the monoamine hypothesis has been further oversimplified when presented to the general public.
Receptor Binding
As of 2012, efforts to determine differences in neurotransmitter receptor expression or for function in the brains of people with MDD using positron emission tomography (PET) had shown inconsistent results. Using the PET imaging technology and reagents available as of 2012, it appeared that the D1 receptor may be under-expressed in the striatum of people with MDD. 5-HT1A receptor binding literature is inconsistent; however, it leans towards a general decrease in the mesiotemporal cortex. 5-HT2A receptor binding appears to be unregulated in people with MDD. Results from studies on 5-HTT binding are variable, but tend to indicate higher levels in people with MDD. Results with D2/D3 receptor binding studies are too inconsistent to draw any conclusions. Evidence supports increased MAO activity in people with MDD, and it may even be a trait marker (not changed by response to treatment). Muscarinic receptor binding appears to be increased in depression, and, given ligand binding dynamics, suggests increased cholinergic activity.
Four meta analyses on receptor binding in depression have been performed, two on serotonin transporter (5-HTT), one on 5-HT1A, and another on dopamine transporter (DAT). One meta analysis on 5-HTT reported that binding was reduced in the midbrain and amygdala, with the former correlating with greater age, and the latter correlating with depression severity. Another meta-analysis on 5-HTT including both post-mortem and in vivo receptor binding studies reported that while in vivo studies found reduced 5-HTT in the striatum, amygdala and midbrain, post mortem studies found no significant associations. 5-HT1A was found to be reduced in the anterior cingulate cortex, mesiotemporal lobe, insula, and hippocampus, but not in the amygdala or occipital lobe. The most commonly used 5-HT1A ligands are not displaced by endogenous serotonin, indicating that receptor density or affinity is reduced. Dopamine transporter binding is not changed in depression.
Emotional Processing and Neural Circuits
Emotional Bias
People with MDD show a number of biases in emotional processing, such as a tendency to rate happy faces more negatively, and a tendency to allocate more attentional resources to sad expressions. Depressed people also have impaired recognition of happy, angry, disgusted, fearful and surprised, but not sad faces. Functional neuroimaging has demonstrated hyperactivity of various brain regions in response to negative emotional stimuli, and hypoactivity in response to positive stimuli. One meta analysis reported that depressed subjects showed decreased activity in the left dorsolateral prefrontal cortex and increased activity in the amygdala in response to negative stimuli. Another meta analysis reported elevated hippocampus and thalamus activity in a subgroup of depressed subjects who were medication naïve, not elderly, and had no comorbidities. The therapeutic lag of antidepressants has been suggested to be a result of antidepressants modifying emotional processing leading to mood changes. This is supported by the observation that both acute and sub-chronic SSRI administration increases response to positive faces. Antidepressant treatment appears to reverse mood congruent biases in limbic, prefrontal, and fusiform areas. dlPFC response is enhanced and amygdala response is attenuated during processing of negative emotions, the former or which is thought to reflect increased top down regulation. The fusiform gyrus and other visual processing areas respond more strongly to positive stimuli with antidepressant treatment, which is thought to reflect the a positive processing bias. These effects do not appear to be unique to serotonergic or noradrenergic antidepressants, but also occur in other forms of treatment such as deep brain stimulation.
Neural Circuits
One meta analysis of functional neuroimaging in depression observed a pattern of abnormal neural activity hypothesized to reflect an emotional processing bias. Relative to controls, people with MDD showed hyperactivity of circuits in the salience network (SN), composed of the pulvinar nuclei, the insula, and the dorsal anterior cingulate cortex (dACC), as well as decreased activity in regulatory circuits composed of the striatum and dlPFC.
A neuroanatomical model called the limbic-cortical model has been proposed to explain early biological findings in depression. The model attempts to relate specific symptoms of depression to neurological abnormalities. Elevated resting amygdala activity was proposed to underlie rumination, as stimulation of the amygdala has been reported to be associated with the intrusive recall of negative memories. The ACC was divided into pregenual (pgACC) and subgenual regions (sgACC), with the former being electrophysiologically associated with fear, and the latter being metabolically implicated in sadness in healthy subjects. Hyperactivity of the lateral orbitofrontal and insular regions, along with abnormalities in lateral prefrontal regions was suggested to underlie maladaptive emotional responses, given the regions roles in reward learning. This model and another termed “the cortical striatal model”, which focused more on abnormalities in the cortico-basal ganglia-thalamo-cortical loop, have been supported by recent literature. Reduced striatal activity, elevated OFC activity, and elevated sgACC activity were all findings consistent with the proposed models. However, amygdala activity was reported to be decreased, contrary to the limbic-cortical model. Furthermore, only lateral prefrontal regions were modulated by treatment, indicating that prefrontal areas are state markers (i.e. dependent upon mood), while subcortical abnormalities are trait markers (i.e. reflect a susceptibility).
Reward
While depression severity as a whole is not correlated with a blunted neural response to reward, anhedonia is directly correlated to reduced activity in the reward system. The study of reward in depression is limited by heterogeneity in the definition and conceptualisations of reward and anhedonia. Anhedonia is broadly defined as a reduced ability to feel pleasure, but questionnaires and clinical assessments rarely distinguish between motivational “wanting” and consummatory “liking”. While a number of studies suggest that depressed subjects rate positive stimuli less positively and as less arousing, a number of studies fail to find a difference. Furthermore, response to natural rewards such as sucrose does not appear to be attenuated. General affective blunting may explain “anhedonic” symptoms in depression, as meta analysis of both positive and negative stimuli reveal reduced rating of intensity. As anhedonia is a prominent symptom of depression, direct comparison of depressed with healthy subjects reveals increased activation of the subgenual anterior cingulate cortex (sgACC), and reduced activation of the ventral striatum, and in particular the nucleus accumbens (NAcc) in response to positive stimuli. Although the finding of reduced NAcc activity during reward paradigms is fairly consistent, the NAcc is made up of a functionally diverse range of neurons, and reduced blood-oxygen-level dependent (BOLD) signal in this region could indicate a variety of things including reduced afferent activity or reduced inhibitory output. Nevertheless, these regions are important in reward processing, and dysfunction of them in depression is thought to underlie anhedonia. Residual anhedonia that is not well targeted by serotonergic antidepressants is hypothesized to result from inhibition of dopamine release by activation of 5-HT2C receptors in the striatum. The response to reward in the medial orbitofrontal cortex (OFC) is attenuated in depression, while lateral OFC response is enhanced to punishment. The lateral OFC shows sustained response to absence of reward or punishment, and it is thought to be necessary for modifying behaviour in response to changing contingencies. Hypersensitivity in the lOFC may lead to depression by producing a similar effect to learned helplessness in animals.
Elevated response in the sgACC is a consistent finding in neuroimaging studies using a number of paradigms including reward related tasks. Treatment is also associated with attenuated activity in the sgACC, and inhibition of neurons in the rodent homologue of the sgACC, the infralimbic cortex (IL), produces an antidepressant effect. Hyperactivity of the sgACC has been hypothesized to lead to depression via attenuating the somatic response to reward or positive stimuli. Contrary to studies of functional magnetic resonance imaging response in the sgACC during tasks, resting metabolism is reduced in the sgACC. However, this is only apparent when correcting for the prominent reduction in sgACC volume associated with depression; structural abnormalities are evident at a cellular level, as neuropathological studies report reduced sgACC cell markers. The model of depression proposed from these findings by Drevets et al. suggests that reduced sgACC activity results in enhanced sympathetic nervous system activity and blunted HPA axis feedback. Activity in the sgACC may also not be causal in depression, as the authors of one review that examined neuroimaging in depressed subjects during emotional regulation hypothesized that the pattern of elevated sgACC activity reflected increased need to modulate automatic emotional responses in depression. More extensive sgACC and general prefrontal recruitment during positive emotional processing was associated with blunted subcortical response to positive emotions, and subject anhedonia. This was interpreted by the authors to reflect a downregulation of positive emotions by the excessive recruitment of the prefrontal cortex.
Neuroanatomy
While a number of neuroimaging findings are consistently reported in people with major depressive disorder, the heterogeneity of depressed populations presents difficulties interpreting these findings. For example, averaging across populations may hide certain subgroup related findings; while reduced dlPFC activity is reported in depression, a subgroup may present with elevated dlPFC activity. Averaging may also yield statistically significant findings, such as reduced hippocampal volumes, that are actually present in a subgroup of subjects. Due to these issues and others, including the longitudinal consistency of depression, most neural models are likely inapplicable to all depression.
Structural Neuroimaging
Meta analyses performed using seed-based d mapping have reported grey matter reductions in a number of frontal regions. One meta analysis of early onset general depression reported grey matter reductions in the bilateral anterior cingulate cortex (ACC) and dorsomedial prefrontal cortex (dmPFC). One meta analysis on first episode depression observed distinct patterns of grey matter reductions in medication free, and combined populations; medication free depression was associated with reductions in the right dorsolateral prefrontal cortex, right amygdala, and right inferior temporal gyrus; analysis on a combination of medication free and medicated depression found reductions in the left insula, right supplementary motor area, and right middle temporal gyrus. Another review distinguishing medicated and medication free populations, albeit not restricted to people with their first episode of MDD, found reductions in the combined population in the bilateral superior, right middle, and left inferior frontal gyrus, along with the bilateral parahippocampus. Increases in thalamic and ACC grey matter was reported in the medication free and medicated populations respectively. A meta analysis performed using “activation likelihood estimate” reported reductions in the paracingulate cortex, dACC and amygdala.
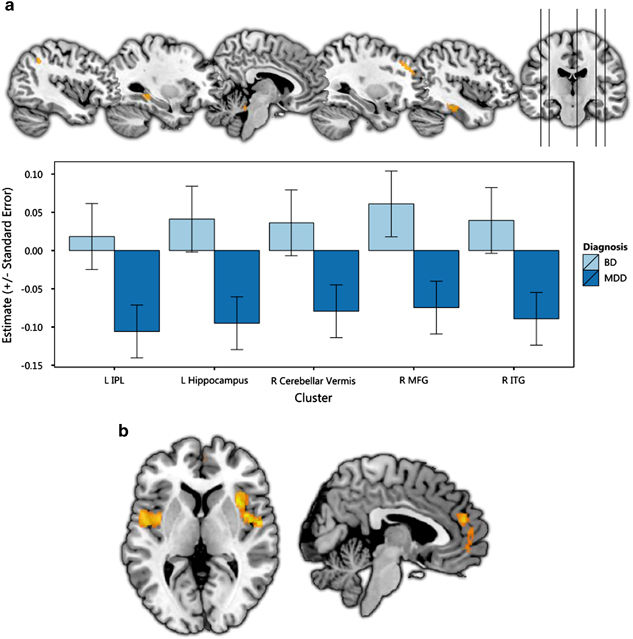
Using statistical parametric mapping, one meta analysis replicated previous findings of reduced grey matter in the ACC, medial prefrontal cortex, inferior frontal gyrus, hippocampus and thalamus; however reductions in the OFC and ventromedial prefrontal cortex grey matter were also reported.
Two studies on depression from the ENIGMA consortium have been published, one on cortical thickness, and the other on subcortical volume. Reduced cortical thickness was reported in the bilateral OFC, ACC, insula, middle temporal gyri, fusiform gyri, and posterior cingulate cortices, while surface area deficits were found in medial occipital, inferior parietal, orbitofrontal and precentral regions. Subcortical abnormalities, including reductions in hippocampus and amygdala volumes, which were especially pronounced in early onset depression.
Multiple meta analysis have been performed on studies assessing white matter integrity using fractional anisotropy (FA). Reduced FA has been reported in the corpus callosum (CC) in both first episode medication naïve, and general major depressive populations. The extent of CC reductions differs from study to study. People with MDD who have not taken antidepressants before have been reported to have reductions only in the body of the CC and only in the genu of the CC. On the other hand, general MDD samples have been reported to have reductions in the body of the CC, the body and genu of the CC, and only the genu of the CC. Reductions of FA have also been reported in the anterior limb of the internal capsule (ALIC) and superior longitudinal fasciculus.
Functional Neuroimaging
Studies of resting state activity have utilised a number of indicators of resting state activity, including regional homogeneity (ReHO), amplitude of low frequency fluctuations (ALFF), fractional amplitude of low frequency fluctuations (fALFF), arterial spin labelling (ASL), and positron emission tomography measures of regional cerebral blood flow or metabolism.

Studies using ALFF and fALFF have reported elevations in ACC activity, with the former primarily reporting more ventral findings, and the latter more dorsal findings. A conjunction analysis of ALFF and CBF studies converged on the left insula, with previously untreated people having increased insula activity. Elevated caudate CBF was also reported A meta analysis combining multiple indicators of resting activity reported elevated anterior cingulate, striatal, and thalamic activity and reduced left insula, post-central gyrus and fusiform gyrus activity. An activation likelihood estimate (ALE) meta analysis of PET/SPECT resting state studies reported reduced activity in the left insula, pregenual and dorsal anterior cingulate cortex and elevated activity in the thalamus, caudate, anterior hippocampus and amygdala. Compared to the ALE meta analysis of PET/SPECT studies, a study using multi-kernel density analysis reported hyperactivity only in the pulvinar nuclei of the thalamus.
Brain Regions
Research on the brains of people with MDD usually shows disturbed patterns of interaction between multiple parts of the brain. Several areas of the brain are implicated in studies seeking to more fully understand the biology of depression:
Subgenual Cingulate
Studies have shown that Brodmann area 25, also known as subgenual cingulate, is metabolically overactive in treatment-resistant depression. This region is extremely rich in serotonin transporters and is considered as a governor for a vast network involving areas like hypothalamus and brain stem, which influences changes in appetite and sleep; the amygdala and insula, which affect the mood and anxiety; the hippocampus, which plays an important role in memory formation; and some parts of the frontal cortex responsible for self-esteem. Thus disturbances in this area or a smaller than normal size of this area contributes to depression. Deep brain stimulation has been targeted to this region in order to reduce its activity in people with treatment resistant depression.
Prefrontal Cortex
One review reported hypoactivity in the prefrontal cortex of those with depression compared to controls. The prefrontal cortex is involved in emotional processing and regulation, and dysfunction of this process may be involved in the aetiology of depression. One study on antidepressant treatment found an increase in PFC activity in response to administration of antidepressants. One meta analysis published in 2012 found that areas of the prefrontal cortex were hypoactive in response to negative stimuli in people with MDD. One study suggested that areas of the prefrontal cortex are part of a network of regions including dorsal and pregenual cingulate, bilateral middle frontal gyrus, insula and superior temporal gyrus that appear to be hypoactive in people with MDD. However the authors cautioned that the exclusion criteria, lack of consistency and small samples limit results.
Amygdala
The amygdala, a structure involved in emotional processing appears to be hyperactive in those with major depressive disorder. The amygdala in unmedicated depressed persons tended to be smaller than in those that were medicated, however aggregate data shows no difference between depressed and healthy persons. During emotional processing tasks right amygdala is more active than the left, however there is no differences during cognitive tasks, and at rest only the left amygdala appears to be more hyperactive. One study, however, found no difference in amygdala activity during emotional processing tasks.
Hippocampus
Atrophy of the hippocampus has been observed during depression, consistent with animal models of stress and neurogenesis.
Stress can cause depression and depression-like symptoms through monoaminergic changes in several key brain regions as well as suppression in hippocampal neurogenesis. This leads to alteration in emotion and cognition related brain regions as well as HPA axis dysfunction. Through the dysfunction, the effects of stress can be exacerbated including its effects on 5-HT. Furthermore, some of these effects are reversed by antidepressant action, which may act by increasing hippocampal neurogenesis. This leads to a restoration in HPA activity and stress reactivity, thus restoring the deleterious effects induced by stress on 5-HT.
The hypothalamic-pituitary-adrenal axis is a chain of endocrine structures that are activated during the body’s response to stressors of various sorts. The HPA axis involves three structure, the hypothalamus which release CRH that stimulates the pituitary gland to release ACTH which stimulates the adrenal glands to release cortisol. Cortisol has a negative feedback effect on the pituitary gland and hypothalamus. In people with MDD this often shows increased activation in depressed people, but the mechanism behind this is not yet known. Increased basal cortisol levels and abnormal response to dexamethasone challenges have been observed in people with MDD. Early life stress has been hypothesized as a potential cause of HPA dysfunction. HPA axis regulation may be examined through a dexamethasone suppression tests, which tests the feedback mechanisms. Non-suppression of dexamethasone is a common finding in depression, but is not consistent enough to be used as a diagnostic tool. HPA axis changes may be responsible for some of the changes such as decreased bone mineral density and increased weight found in people with MDD. One drug, ketoconazole, currently under development has shown promise in treating MDD.
Hippocampal Neurogenesis
Reduced hippocampal neurogenesis leads to a reduction in hippocampal volume. A genetically smaller hippocampus has been linked to a reduced ability to process psychological trauma and external stress, and subsequent predisposition to psychological illness. Depression without familial risk or childhood trauma has been linked to a normal hippocampal volume but localised dysfunction.
Animal Models
A number of animal models exist for depression, but they are limited in that depression involves primarily subjective emotional changes. However, some of these changes are reflected in physiology and behaviour, the latter of which is the target of many animal models. These models are generally assessed according to four facets of validity; the reflection of the core symptoms in the model; the predictive validity of the model; the validity of the model with regard to human characteristics of aetiology; and the biological plausibility.
Different models for inducing depressive behaviours have been utilised; neuroanatomical manipulations such as olfactory bulbectomy or circuit specific manipulations with optogenetics; genetic models such as 5-HT1A knockout or selectively bred animals; models involving environmental manipulation associated with depression in humans, including chronic mild stress, early life stress and learned helplessness. The validity of these models in producing depressive behaviours may be assessed with a number of behavioural tests. Anhedonia and motivational deficits may, for example, be assessed via examining an animal’s level of engagement with rewarding stimuli such as sucrose or intracranial self-stimulation. Anxious and irritable symptoms may be assessed with exploratory behaviour in the presence of a stressful or novelty environment, such as the open field test, novelty suppressed feeding, or the elevated plus-maze. Fatigue, psychomotor poverty, and agitation may be assessed with locomotor activity, grooming activity, and open field tests.
Animal models possess a number of limitations due to the nature of depression. Some core symptoms of depression, such as rumination, low self-esteem, guilt, and depressed mood cannot be assessed in animals as they require subjective reporting. From an evolutionary standpoint, the behaviour correlates of defeats of loss are thought to be an adaptive response to prevent further loss. Therefore, attempts to model depression that seeks to induce defeat or despair may actually reflect adaption and not disease. Furthermore, while depression and anxiety are frequently comorbid, dissociation of the two in animal models is difficult to achieve. Pharmacological assessment of validity is frequently disconnected from clinical pharmacotherapeutics in that most screening tests assess acute effects, while antidepressants normally take a few weeks to work in humans.
Neurocircuits
Regions involved in reward are common targets of manipulation in animal models of depression, including the nucleus accumbens (NAc), ventral tegmental area (VTA), ventral pallidum (VP), lateral habenula (LHb) and medial prefrontal cortex (mPFC). Tentative fMRI studies in humans demonstrate elevated LHb activity in depression. The lateral habenula projects to the RMTg to drive inhibition of dopamine neurons in the VTA during omission of reward. In animal models of depression, elevated activity has been reported in LHb neurons that project to the ventral tegmental area (ostensibly reducing dopamine release). The LHb also projects to aversion reactive mPFC neurons, which may provide an indirect mechanism for producing depressive behaviours. Learned helplessness induced potentiation of LHb synapses are reversed by antidepressant treatment, providing predictive validity. A number of inputs to the LHb have been implicated in producing depressive behaviours. Silencing GABAergic projections from the NAc to the LHb reduces conditioned place preference induced in social aggression, and activation of these terminals induces CPP. Ventral pallidum firing is also elevated by stress induced depression, an effect that is pharmacologically valid, and silencing of these neurons alleviates behavioural correlates of depression. Tentative in vivo evidence from people with MDD suggests abnormalities in dopamine signalling. This led to early studies investigating VTA activity and manipulations in animal models of depression. Massive destruction of VTA neurons enhances depressive behaviours, while VTA neurons reduce firing in response to chronic stress. However, more recent specific manipulations of the VTA produce varying results, with the specific animal model, duration of VTA manipulation, method of VTA manipulation, and subregion of VTA manipulation all potentially leading to differential outcomes. Stress and social defeat induced depressive symptoms, including anhedonia, are associated with potentiation of excitatory inputs to Dopamine D2 receptor-expressing medium spiny neurons (D2-MSNs) and depression of excitatory inputs to Dopamine D1 receptor-expressing medium spiny neurons (D1-MSNs). Optogenetic excitation of D1-MSNs alleviates depressive symptoms and is rewarding, while the same with D2-MSNs enhances depressive symptoms. Excitation of glutaminergic inputs from the ventral hippocampus reduces social interactions, and enhancing these projections produces susceptibility to stress-induced depression. Manipulations of different regions of the mPFC can produce and attenuate depressive behaviours. For example, inhibiting mPFC neurons specifically in the intralimbic cortex attenuates depressive behaviours. The conflicting findings associated with mPFC stimulation, when compared to the relatively specific findings in the infralimbic cortex, suggest that the prelimbic cortex and infralimbic cortex may mediate opposing effects. mPFC projections to the raphe nuclei are largely GABAergic and inhibit the firing of serotonergic neurons. Specific activation of these regions reduce immobility in the forced swim test but do not affect open field or forced swim behaviour. Inhibition of the raphe shifts the behavioural phenotype of uncontrolled stress to a phenotype closer to that of controlled stress.
Altered Neuroplasticity
Recent studies have called attention to the role of altered neuroplasticity in depression. A review found a convergence of three phenomena:
- Chronic stress reduces synaptic and dendritic plasticity;
- Depressed subjects show evidence of impaired neuroplasticity (e.g. shortening and reduced complexity of dendritic trees); and
- Anti-depressant medications may enhance neuroplasticity at both a molecular and dendritic level.
The conclusion is that disrupted neuroplasticity is an underlying feature of depression, and is reversed by antidepressants.
Blood levels of BDNF in people with MDD increase significantly with antidepressant treatment and correlate with decrease in symptoms. Post mortem studies and rat models demonstrate decreased neuronal density in the prefrontal cortex thickness in people with MDD. Rat models demonstrate histological changes consistent with MRI findings in humans, however studies on neurogenesis in humans are limited. Antidepressants appear to reverse the changes in neurogenesis in both animal models and humans.
Inflammation
Various reviews have found that general inflammation may play a role in depression. One meta analysis of cytokines in people with MDD found increased levels of pro-inflammatory IL-6 and TNF-a levels relative to controls. The first theories came about when it was noticed that interferon therapy caused depression in a large number of people receiving it. Meta analysis on cytokine levels in people with MDD have demonstrated increased levels of IL-1, IL-6, C-reactive protein, but not IL-10. Increased numbers of T-Cells presenting activation markers, levels of neopterin, IFN gamma, sTNFR, and IL-2 receptors have been observed in depression. Various sources of inflammation in depressive illness have been hypothesized and include trauma, sleep problems, diet, smoking and obesity. Cytokines, by manipulating neurotransmitters, are involved in the generation of sickness behaviour, which shares some overlap with the symptoms of depression. Neurotransmitters hypothesized to be affected include dopamine and serotonin, which are common targets for antidepressant drugs. Induction of indolamine-2,3 dioxygenease by cytokines has been proposed as a mechanism by which immune dysfunction causes depression. One review found normalization of cytokine levels after successful treatment of depression. A meta analysis published in 2014 found the use of anti-inflammatory drugs such as NSAIDs and investigational cytokine inhibitors reduced depressive symptoms. Exercise can act as a stressor, decreasing the levels of IL-6 and TNF-a and increasing those of IL-10, an anti-inflammatory cytokine.
Inflammation is also intimately linked with metabolic processes in humans. For example, low levels of Vitamin D have been associated with greater risk for depression. The role of metabolic biomarkers in depression is an active research area. Recent work has explored the potential relationship between plasma sterols and depressive symptom severity.
Oxidative Stress
A marker of DNA oxidation, 8-Oxo-2′-deoxyguanosine, has been found to be increased in both the plasma and urine of people with MDD. This along with the finding of increased F2-isoprostanes levels found in blood, urine and cerebrospinal fluid indicate increased damage to lipids and DNA in people with MDD. Studies with 8-Oxo-2′ Deoxyguanosine varied by methods of measurement and type of depression, but F2-Isoprostane level was consistent across depression types. Authors suggested lifestyle factors, dysregulation of the HPA axis, immune system and autonomics nervous system as possible causes. Another meta-analysis found similar results with regards to oxidative damage products as well as decreased oxidative capacity. Oxidative DNA damage may play a role in MDD.
Mitochondrial Dysfunction:
Increased markers of oxidative stress relative to controls have been found in people with MDD. These markers include high levels of RNS and ROS which have been shown to influence chronic inflammation, damaging the electron transport chain and biochemical cascades in mitochondria. This lowers the activity of enzymes in the respiratory chain resulting in mitochondrial dysfunction. The brain is a highly energy-consuming and has little capacity to store glucose as glycogen and so depends greatly on mitochondria. Mitochondrial dysfunction has been linked to the dampened neuroplasticity observed in depressed brains.
Large-Scale Brain Network Theory
Instead of studying one brain region, studying large scale brain networks is another approach to understanding psychiatric and neurological disorders, supported by recent research that has shown that multiple brain regions are involved in these disorders. Understanding the disruptions in these networks may provide important insights into interventions for treating these disorders. Recent work suggests that at least three large-scale brain networks are important in psychopathology.
Central Executive Network
The central executive network is made up of fronto-parietal regions, including dorsolateral prefrontal cortex and lateral posterior parietal cortex. This network is involved in high level cognitive functions such as maintaining and using information in working memory, problem solving, and decision making. Deficiencies in this network are common in most major psychiatric and neurological disorders, including depression. Because this network is crucial for everyday life activities, those who are depressed can show impairment in basic activities like test taking and being decisive.
Default Mode Network
The default mode network includes hubs in the prefrontal cortex and posterior cingulate, with other prominent regions of the network in the medial temporal lobe and angular gyrus. The default mode network is usually active during mind-wandering and thinking about social situations. In contrast, during specific tasks probed in cognitive science (for example, simple attention tasks), the default network is often deactivated. Research has shown that regions in the default mode network (including medial prefrontal cortex and posterior cingulate) show greater activity when depressed participants ruminate (that is, when they engage in repetitive self-focused thinking) than when typical, healthy participants ruminate. People with MDD also show increased connectivity between the default mode network and the subgenual cingulate and the adjoining ventromedial prefrontal cortex in comparison to healthy individuals, individuals with dementia or with autism. Numerous studies suggest that the subgenual cingulate plays an important role in the dysfunction that characterizes major depression. The increased activation in the default mode network during rumination and the atypical connectivity between core default mode regions and the subgenual cingulate may underlie the tendency for depressed individual to get “stuck” in the negative, self-focused thoughts that often characterise depression. However, further research is needed to gain a precise understanding of how these network interactions map to specific symptoms of depression.
Salience Network
The salience network is a cingulate-frontal operculum network that includes core nodes in the anterior cingulate and anterior insula. A salience network is a large-scale brain network involved in detecting and orienting the most pertinent of the external stimuli and internal events being presented. Individuals who have a tendency to experience negative emotional states (scoring high on measures of neuroticism) show an increase in the right anterior insula during decision-making, even if the decision has already been made. This atypically high activity in the right anterior insula is thought to contribute to the experience of negative and worrisome feelings. In MDD, anxiety is often a part of the emotional state that characterises depression.
