The Brief Psychiatric Rating Scale (BPRS) is a rating scale which a clinician or researcher may use to measure psychiatric symptoms such as depression, anxiety, hallucinations and unusual behaviour.
The scale is one of the oldest, most widely used scales to measure psychotic symptoms and was first published in 1962.
Brief History
The BPRS was initially developed by John E. Overall and Donald R. Gorham. It was created for the purpose of being able to quickly assess the patient’s psychiatric symptoms prior, during, or following a treatment. The items of the test were generated from conducting factor analysis on the Multidimensional Scale for Rating Psychiatric Patients and the Inpatient Multidimensional Psychiatric Scale. Sixteen factors were found from the analysis, which served as the building blocks for the BPRS. Later research in 1968 added two more factors to the BPRS, which were excitement and disorientation.
Test Format
The BPRS consists of 18 items measuring the following factors:
It uses a seven-item Likert scale with the following values:
1 = “not present”.
2 = “very mild”.
3 = “mild”.
4 = “moderate”.
5 = “moderately severe”.
6 = “severe”.
7 = “extremely severe”.
The test is administered in tandem with a series of interviews conducted by at least two clinicians to ensure interrater reliability of the assessment.
Usage
The BPRS is intended for use on adult psychiatric patients and has been validated for use in elderly populations. A version designed for children called the Brief Psychiatric Rating Scale Children was also developed by Overall and Betty Pfeifferbaum, with different scale structures and factors.
Further Development
An expanded version of the test was created in 1993 by D. Lukoff, Keith H. Nuechterlein, and Joseph Ventura.
This page is based on the copyrighted Wikipedia article <https://en.wikipedia.org/wiki/Brief_Psychiatric_Rating_Scale>; it is used under the Creative Commons Attribution-ShareAlike 3.0 Unported License (CC-BY-SA). You may redistribute it, verbatim or modified, providing that you comply with the terms of the CC-BY-SA.
Scientific studies have found that different brain areas show altered activity in people with major depressive disorder (MDD), and this has encouraged advocates of various theories that seek to identify a biochemical origin of the disease, as opposed to theories that emphasize psychological or situational causes.
Factors spanning these causative groups include nutritional deficiencies in magnesium, vitamin D, and tryptophan with situational origin but biological impact. Several theories concerning the biologically based cause of depression have been suggested over the years, including theories revolving around monoamine neurotransmitters, neuroplasticity, neurogenesis, inflammation and the circadian rhythm. Physical illnesses, including hypothyroidism and mitochondrial disease, can also trigger depressive symptoms.
Neural circuits implicated in depression include those involved in the generation and regulation of emotion, as well as in reward. Abnormalities are commonly found in the lateral prefrontal cortex whose putative function is generally considered to involve regulation of emotion. Regions involved in the generation of emotion and reward such as the amygdala, anterior cingulate cortex (ACC), orbitofrontal cortex (OFC), and striatum are frequently implicated as well. These regions are innervated by a monoaminergic nuclei, and tentative evidence suggests a potential role for abnormal monoaminergic activity.
Genetic Factors
Difficulty of Gene Studies
Historically, candidate gene studies have been a major focus of study. However, as the number of genes reduces the likelihood of choosing a correct candidate gene, Type I errors (false positives) are highly likely. Candidate genes studies frequently possess a number of flaws, including frequent genotyping errors and being statistically underpowered. These effects are compounded by the usual assessment of genes without regard for gene-gene interactions. These limitations are reflected in the fact that no candidate gene has reached genome-wide significance.
Gene Candidates
5-HTTLPR
The 5-HTTLPR, or serotonin transporter promoter gene’s short allele, has been associated with increased risk of depression; since the 1990s, however, results have been inconsistent. Other genes that have been linked to a gene-environment interaction include CRHR1, FKBP5 and BDNF, the first two of which are related to the stress reaction of the HPA axis, and the latter of which is involved in neurogenesis. Candidate gene analysis of 5-HTTLPR on depression was inconclusive on its effect, either alone or in combination with life stress.
A 2003 study proposed that a gene-environment interaction (GxE) may explain why life stress is a predictor for depressive episodes in some individuals, but not in others, depending on an allelic variation of the serotonin-transporter-linked promoter region (5-HTTLPR). This hypothesis was widely-discussed in both the scientific literature and popular media, where it was dubbed the “Orchid gene”, but has conclusively failed to replicate in much larger samples, and the observed effect sizes in earlier work are not consistent with the observed polygenicity of depression.
BDNF
BDNF polymorphisms have also been hypothesized to have a genetic influence, but early findings and research failed to replicate in larger samples, and the effect sizes found by earlier estimates are inconsistent with the observed polygenicity of depression.
SIRT1 and LHPP
A 2015 GWAS study in Han Chinese women positively identified two variants in intronic regions near SIRT1 and LHPP with a genome-wide significant association.
Norepinephrine Transporter Polymorphisms
Attempts to find a correlation between norepinephrine transporter polymorphisms and depression have yielded negative results.
One review identified multiple frequently studied candidate genes. The genes encoding for the 5-HTT and 5-HT2A receptor were inconsistently associated with depression and treatment response. Mixed results were found for brain-derived neurotrophic factor (BDNF) Val66Met polymorphisms. Polymorphisms in the tryptophan hydroxylase gene was found to be tentatively associated with suicidal behaviour. A meta analysis of 182 case controlled genetic studies published in 2008 found Apolipoprotein E verepsilon 2 to be protective, and GNB3 825T, MTHFR 677T, SLC6A4 44bp insertion or deletions, and SLC6A3 40 bpVNTR 9/10 genotype to confer risk.
Circadian Rhythm
Depression may be related to abnormalities in the circadian rhythm, or biological clock.
A well synchronised circadian rhythm is critical for maintaining optimal health. Adverse changes and alterations in the circadian rhythm have been associated various neurological disorders and mood disorders including depression.
Depression may be related to the same brain mechanisms that control the cycles of sleep and wakefulness.
Sleep
Sleep disturbance is the most prominent symptom in depressive patients. Studies about sleep electroencephalograms have shown characteristic changes in depression such as reductions in non-rapid eye movement sleep production, disruptions of sleep continuity and disinhibition of rapid eye movement (REM) sleep. Rapid eye movement (REM) sleep – the stage in which dreaming occurs – may be quick to arrive and intense in depressed people. REM sleep depends on decreased serotonin levels in the brain stem, and is impaired by compounds, such as antidepressants, that increase serotonergic tone in brain stem structures. Overall, the serotonergic system is least active during sleep and most active during wakefulness. Prolonged wakefulness due to sleep deprivation activates serotonergic neurons, leading to processes similar to the therapeutic effect of antidepressants, such as the selective serotonin reuptake inhibitors (SSRIs). Depressed individuals can exhibit a significant lift in mood after a night of sleep deprivation. SSRIs may directly depend on the increase of central serotonergic neurotransmission for their therapeutic effect, the same system that impacts cycles of sleep and wakefulness.
Light Therapy
Research on the effects of light therapy on seasonal affective disorder suggests that light deprivation is related to decreased activity in the serotonergic system and to abnormalities in the sleep cycle, particularly insomnia. Exposure to light also targets the serotonergic system, providing more support for the important role this system may play in depression. Sleep deprivation and light therapy both target the same brain neurotransmitter system and brain areas as antidepressant drugs, and are now used clinically to treat depression. Light therapy, sleep deprivation and sleep time displacement (sleep phase advance therapy) are being used in combination quickly to interrupt a deep depression in people who are hospitalised for MDD.
Increased and decreased sleep length appears to be a risk factor for depression. People with MDD sometimes show diurnal and seasonal variation of symptom severity, even in non-seasonal depression. Diurnal mood improvement was associated with activity of dorsal neural networks. Increased mean core temperature was also observed. One hypothesis proposed that depression was a result of a phase shift.
Daytime light exposure correlates with decreased serotonin transporter activity, which may underlie the seasonality of some depression.
Monoamines
Monoamines are neurotransmitters that include serotonin, dopamine, norepinephrine, and epinephrine.
Illustration of the major elements in a prototypical synapse. Synapses are gaps between nerve cells. These cells convert their electrical impulses into bursts of chemical relayers, called neurotransmitters, which travel across the synapses to receptors on adjacent cells, triggering electrical impulses to travel down the latter cells.
Monoamine Hypothesis of Depression
Many antidepressant drugs acutely increase synaptic levels of the monoamine neurotransmitter, serotonin, but they may also enhance the levels of norepinephrine and dopamine. The observation of this efficacy led to the monoamine hypothesis of depression, which postulates that the deficit of certain neurotransmitters is responsible for depression, and even that certain neurotransmitters are linked to specific symptoms. Normal serotonin levels have been linked to mood and behaviour regulation, sleep, and digestion; norepinephrine to the fight-or-flight response; and dopamine to movement, pleasure, and motivation. Some have also proposed the relationship between monoamines and phenotypes such as serotonin in sleep and suicide, norepinephrine in dysphoria, fatigue, apathy, cognitive dysfunction, and dopamine in loss of motivation and psychomotor symptoms.[31] The main limitation for the monoamine hypothesis of depression is the therapeutic lag between initiation of antidepressant treatment and perceived improvement of symptoms. One explanation for this therapeutic lag is that the initial increase in synaptic serotonin is only temporary, as firing of serotonergic neurons in the dorsal raphe adapt via the activity of 5-HT1A autoreceptors. The therapeutic effect of antidepressants is thought to arise from autoreceptor desensitization over a period of time, eventually elevating firing of serotonergic neurons.
Serotonin
Initial studies of serotonin in depression examined peripheral measures such as the serotonin metabolite 5-Hydroxyindoleacetic acid (5-HIAA) and platelet binding. The results were generally inconsistent, and may not generalise to the central nervous system. However evidence from receptor binding studies and pharmacological challenges provide some evidence for dysfunction of serotonin neurotransmission in depression. Serotonin may indirectly influence mood by altering emotional processing biases that are seen at both the cognitive/behavioural and neural level. Pharmacologically reducing serotonin synthesis, and pharmacologically enhancing synaptic serotonin can produce and attenuate negative affective biases, respectively. These emotional processing biases may explain the therapeutic gap.
Dopamine
While various abnormalities have been observed in dopaminergic systems, results have been inconsistent. People with MDD have an increased reward response to dextroamphetamine compared to controls, and it has been suggested that this results from hypersensitivity of dopaminergic pathways due to natural hypoactivity. While polymorphisms of the D4 and D3 receptor have been implicated in depression, associations have not been consistently replicated. Similar inconsistency has been found in post-mortem studies, but various dopamine receptor agonists show promise in treating MDD. There is some evidence that there is decreased nigrostriatal pathway activity in people with melancholic depression (psychomotor retardation). Further supporting the role of dopamine in depression is the consistent finding of decreased cerebrospinal fluid and jugular metabolites of dopamine, as well as post mortem findings of altered Dopamine receptor D3 and dopamine transporter expression. Studies in rodents have supported a potential mechanism involving stress-induced dysfunction of dopaminergic systems.
Monoamine receptors affect phospholipase C and adenylyl cyclase inside of the cell. Green arrows means stimulation and red arrows inhibition. Serotonin receptors are blue, norepinephrine orange, and dopamine yellow. Phospholipase C and adenylyl cyclase start a signalling cascade which turn on or off genes in the cell. Sufficient ATP from mitochondria is required for these downstream signalling events. The 5HT-3 receptor is associated with gastrointestinal adverse effects and has no relationship to the other monoamine receptors.
Catecholamines
A number of lines of evidence indicative of decreased adrenergic activity in depression have been reported. Findings include the decreased activity of tyrosine hydroxylase, decreased size of the locus coeruleus, increased alpha 2 adrenergic receptor density, and decreased alpha 1 adrenergic receptor density. Furthermore, norepinephrine transporter knockout in mice models increases their tolerance to stress, implicating norepinephrine in depression.
One method used to study the role of monoamines is monoamine depletion. Depletion of tryptophan (the precursor of serotonin), tyrosine and phenylalanine (precursors to dopamine) does result in decreased mood in those with a predisposition to depression, but not in persons lacking the predisposition. On the other hand, inhibition of dopamine and norepinephrine synthesis with alpha-methyl-para-tyrosine does not consistently result in decreased mood.
Monoamine Oxidase
An offshoot of the monoamine hypothesis suggests that monoamine oxidase A (MAO-A), an enzyme which metabolises monoamines, may be overly active in depressed people. This would, in turn, cause the lowered levels of monoamines. This hypothesis received support from a PET study, which found significantly elevated activity of MAO-A in the brain of some depressed people. In genetic studies, the alterations of MAO-A-related genes have not been consistently associated with depression. Contrary to the assumptions of the monoamine hypothesis, lowered but not heightened activity of MAO-A was associated with depressive symptoms in adolescents. This association was observed only in maltreated youth, indicating that both biological (MAO genes) and psychological (maltreatment) factors are important in the development of depressive disorders. In addition, some evidence indicates that disrupted information processing within neural networks, rather than changes in chemical balance, might underlie depression.
Limitations
Since the 1990s, research has uncovered multiple limitations of the monoamine hypothesis, and its inadequacy has been criticised within the psychiatric community. For one thing, serotonin system dysfunction cannot be the sole cause of depression. Not all patients treated with antidepressants show improvements despite the usually rapid increase in synaptic serotonin. If significant mood improvements do occur, this is often not for at least two to four weeks. One possible explanation for this lag is that the neurotransmitter activity enhancement is the result of auto receptor desensitization, which can take weeks. Intensive investigation has failed to find convincing evidence of a primary dysfunction of a specific monoamine system in people with MDD. The antidepressants that do not act through the monoamine system, such as tianeptine and opipramol, have been known for a long time. There have also been inconsistent findings with regard to levels of serum 5-HIAA, a metabolite of serotonin. Experiments with pharmacological agents that cause depletion of monoamines have shown that this depletion does not cause depression in healthy people. Another problem that presents is that drugs that deplete monoamines may actually have antidepressant properties. Further, some have argued that depression may be marked by a hyperserotonergic state. Already limited, the monoamine hypothesis has been further oversimplified when presented to the general public.
Receptor Binding
As of 2012, efforts to determine differences in neurotransmitter receptor expression or for function in the brains of people with MDD using positron emission tomography (PET) had shown inconsistent results. Using the PET imaging technology and reagents available as of 2012, it appeared that the D1 receptor may be under-expressed in the striatum of people with MDD. 5-HT1A receptor binding literature is inconsistent; however, it leans towards a general decrease in the mesiotemporal cortex. 5-HT2A receptor binding appears to be unregulated in people with MDD. Results from studies on 5-HTT binding are variable, but tend to indicate higher levels in people with MDD. Results with D2/D3 receptor binding studies are too inconsistent to draw any conclusions. Evidence supports increased MAO activity in people with MDD, and it may even be a trait marker (not changed by response to treatment). Muscarinic receptor binding appears to be increased in depression, and, given ligand binding dynamics, suggests increased cholinergic activity.
Four meta analyses on receptor binding in depression have been performed, two on serotonin transporter (5-HTT), one on 5-HT1A, and another on dopamine transporter (DAT). One meta analysis on 5-HTT reported that binding was reduced in the midbrain and amygdala, with the former correlating with greater age, and the latter correlating with depression severity. Another meta-analysis on 5-HTT including both post-mortem and in vivo receptor binding studies reported that while in vivo studies found reduced 5-HTT in the striatum, amygdala and midbrain, post mortem studies found no significant associations. 5-HT1A was found to be reduced in the anterior cingulate cortex, mesiotemporal lobe, insula, and hippocampus, but not in the amygdala or occipital lobe. The most commonly used 5-HT1A ligands are not displaced by endogenous serotonin, indicating that receptor density or affinity is reduced. Dopamine transporter binding is not changed in depression.
Emotional Processing and Neural Circuits
Emotional Bias
People with MDD show a number of biases in emotional processing, such as a tendency to rate happy faces more negatively, and a tendency to allocate more attentional resources to sad expressions. Depressed people also have impaired recognition of happy, angry, disgusted, fearful and surprised, but not sad faces. Functional neuroimaging has demonstrated hyperactivity of various brain regions in response to negative emotional stimuli, and hypoactivity in response to positive stimuli. One meta analysis reported that depressed subjects showed decreased activity in the left dorsolateral prefrontal cortex and increased activity in the amygdala in response to negative stimuli. Another meta analysis reported elevated hippocampus and thalamus activity in a subgroup of depressed subjects who were medication naïve, not elderly, and had no comorbidities. The therapeutic lag of antidepressants has been suggested to be a result of antidepressants modifying emotional processing leading to mood changes. This is supported by the observation that both acute and sub-chronic SSRI administration increases response to positive faces. Antidepressant treatment appears to reverse mood congruent biases in limbic, prefrontal, and fusiform areas. dlPFC response is enhanced and amygdala response is attenuated during processing of negative emotions, the former or which is thought to reflect increased top down regulation. The fusiform gyrus and other visual processing areas respond more strongly to positive stimuli with antidepressant treatment, which is thought to reflect the a positive processing bias. These effects do not appear to be unique to serotonergic or noradrenergic antidepressants, but also occur in other forms of treatment such as deep brain stimulation.
Neural Circuits
One meta analysis of functional neuroimaging in depression observed a pattern of abnormal neural activity hypothesized to reflect an emotional processing bias. Relative to controls, people with MDD showed hyperactivity of circuits in the salience network (SN), composed of the pulvinar nuclei, the insula, and the dorsal anterior cingulate cortex (dACC), as well as decreased activity in regulatory circuits composed of the striatum and dlPFC.
A neuroanatomical model called the limbic-cortical model has been proposed to explain early biological findings in depression. The model attempts to relate specific symptoms of depression to neurological abnormalities. Elevated resting amygdala activity was proposed to underlie rumination, as stimulation of the amygdala has been reported to be associated with the intrusive recall of negative memories. The ACC was divided into pregenual (pgACC) and subgenual regions (sgACC), with the former being electrophysiologically associated with fear, and the latter being metabolically implicated in sadness in healthy subjects. Hyperactivity of the lateral orbitofrontal and insular regions, along with abnormalities in lateral prefrontal regions was suggested to underlie maladaptive emotional responses, given the regions roles in reward learning. This model and another termed “the cortical striatal model”, which focused more on abnormalities in the cortico-basal ganglia-thalamo-cortical loop, have been supported by recent literature. Reduced striatal activity, elevated OFC activity, and elevated sgACC activity were all findings consistent with the proposed models. However, amygdala activity was reported to be decreased, contrary to the limbic-cortical model. Furthermore, only lateral prefrontal regions were modulated by treatment, indicating that prefrontal areas are state markers (i.e. dependent upon mood), while subcortical abnormalities are trait markers (i.e. reflect a susceptibility).
Reward
While depression severity as a whole is not correlated with a blunted neural response to reward, anhedonia is directly correlated to reduced activity in the reward system. The study of reward in depression is limited by heterogeneity in the definition and conceptualisations of reward and anhedonia. Anhedonia is broadly defined as a reduced ability to feel pleasure, but questionnaires and clinical assessments rarely distinguish between motivational “wanting” and consummatory “liking”. While a number of studies suggest that depressed subjects rate positive stimuli less positively and as less arousing, a number of studies fail to find a difference. Furthermore, response to natural rewards such as sucrose does not appear to be attenuated. General affective blunting may explain “anhedonic” symptoms in depression, as meta analysis of both positive and negative stimuli reveal reduced rating of intensity. As anhedonia is a prominent symptom of depression, direct comparison of depressed with healthy subjects reveals increased activation of the subgenual anterior cingulate cortex (sgACC), and reduced activation of the ventral striatum, and in particular the nucleus accumbens (NAcc) in response to positive stimuli. Although the finding of reduced NAcc activity during reward paradigms is fairly consistent, the NAcc is made up of a functionally diverse range of neurons, and reduced blood-oxygen-level dependent (BOLD) signal in this region could indicate a variety of things including reduced afferent activity or reduced inhibitory output. Nevertheless, these regions are important in reward processing, and dysfunction of them in depression is thought to underlie anhedonia. Residual anhedonia that is not well targeted by serotonergic antidepressants is hypothesized to result from inhibition of dopamine release by activation of 5-HT2C receptors in the striatum. The response to reward in the medial orbitofrontal cortex (OFC) is attenuated in depression, while lateral OFC response is enhanced to punishment. The lateral OFC shows sustained response to absence of reward or punishment, and it is thought to be necessary for modifying behaviour in response to changing contingencies. Hypersensitivity in the lOFC may lead to depression by producing a similar effect to learned helplessness in animals.
Elevated response in the sgACC is a consistent finding in neuroimaging studies using a number of paradigms including reward related tasks. Treatment is also associated with attenuated activity in the sgACC, and inhibition of neurons in the rodent homologue of the sgACC, the infralimbic cortex (IL), produces an antidepressant effect. Hyperactivity of the sgACC has been hypothesized to lead to depression via attenuating the somatic response to reward or positive stimuli. Contrary to studies of functional magnetic resonance imaging response in the sgACC during tasks, resting metabolism is reduced in the sgACC. However, this is only apparent when correcting for the prominent reduction in sgACC volume associated with depression; structural abnormalities are evident at a cellular level, as neuropathological studies report reduced sgACC cell markers. The model of depression proposed from these findings by Drevets et al. suggests that reduced sgACC activity results in enhanced sympathetic nervous system activity and blunted HPA axis feedback. Activity in the sgACC may also not be causal in depression, as the authors of one review that examined neuroimaging in depressed subjects during emotional regulation hypothesized that the pattern of elevated sgACC activity reflected increased need to modulate automatic emotional responses in depression. More extensive sgACC and general prefrontal recruitment during positive emotional processing was associated with blunted subcortical response to positive emotions, and subject anhedonia. This was interpreted by the authors to reflect a downregulation of positive emotions by the excessive recruitment of the prefrontal cortex.
Neuroanatomy
While a number of neuroimaging findings are consistently reported in people with major depressive disorder, the heterogeneity of depressed populations presents difficulties interpreting these findings. For example, averaging across populations may hide certain subgroup related findings; while reduced dlPFC activity is reported in depression, a subgroup may present with elevated dlPFC activity. Averaging may also yield statistically significant findings, such as reduced hippocampal volumes, that are actually present in a subgroup of subjects. Due to these issues and others, including the longitudinal consistency of depression, most neural models are likely inapplicable to all depression.
Structural Neuroimaging
Meta analyses performed using seed-based d mapping have reported grey matter reductions in a number of frontal regions. One meta analysis of early onset general depression reported grey matter reductions in the bilateral anterior cingulate cortex (ACC) and dorsomedial prefrontal cortex (dmPFC). One meta analysis on first episode depression observed distinct patterns of grey matter reductions in medication free, and combined populations; medication free depression was associated with reductions in the right dorsolateral prefrontal cortex, right amygdala, and right inferior temporal gyrus; analysis on a combination of medication free and medicated depression found reductions in the left insula, right supplementary motor area, and right middle temporal gyrus. Another review distinguishing medicated and medication free populations, albeit not restricted to people with their first episode of MDD, found reductions in the combined population in the bilateral superior, right middle, and left inferior frontal gyrus, along with the bilateral parahippocampus. Increases in thalamic and ACC grey matter was reported in the medication free and medicated populations respectively. A meta analysis performed using “activation likelihood estimate” reported reductions in the paracingulate cortex, dACC and amygdala.
GMV reductions in MDD and BD.
Using statistical parametric mapping, one meta analysis replicated previous findings of reduced grey matter in the ACC, medial prefrontal cortex, inferior frontal gyrus, hippocampus and thalamus; however reductions in the OFC and ventromedial prefrontal cortex grey matter were also reported.
Two studies on depression from the ENIGMA consortium have been published, one on cortical thickness, and the other on subcortical volume. Reduced cortical thickness was reported in the bilateral OFC, ACC, insula, middle temporal gyri, fusiform gyri, and posterior cingulate cortices, while surface area deficits were found in medial occipital, inferior parietal, orbitofrontal and precentral regions. Subcortical abnormalities, including reductions in hippocampus and amygdala volumes, which were especially pronounced in early onset depression.
Multiple meta analysis have been performed on studies assessing white matter integrity using fractional anisotropy (FA). Reduced FA has been reported in the corpus callosum (CC) in both first episode medication naïve, and general major depressive populations. The extent of CC reductions differs from study to study. People with MDD who have not taken antidepressants before have been reported to have reductions only in the body of the CC and only in the genu of the CC. On the other hand, general MDD samples have been reported to have reductions in the body of the CC, the body and genu of the CC, and only the genu of the CC. Reductions of FA have also been reported in the anterior limb of the internal capsule (ALIC) and superior longitudinal fasciculus.
Functional Neuroimaging
Studies of resting state activity have utilised a number of indicators of resting state activity, including regional homogeneity (ReHO), amplitude of low frequency fluctuations (ALFF), fractional amplitude of low frequency fluctuations (fALFF), arterial spin labelling (ASL), and positron emission tomography measures of regional cerebral blood flow or metabolism.
MDD is associated with reduced FA in the ALIC and genu/body of the CC.
Studies using ALFF and fALFF have reported elevations in ACC activity, with the former primarily reporting more ventral findings, and the latter more dorsal findings. A conjunction analysis of ALFF and CBF studies converged on the left insula, with previously untreated people having increased insula activity. Elevated caudate CBF was also reported A meta analysis combining multiple indicators of resting activity reported elevated anterior cingulate, striatal, and thalamic activity and reduced left insula, post-central gyrus and fusiform gyrus activity. An activation likelihood estimate (ALE) meta analysis of PET/SPECT resting state studies reported reduced activity in the left insula, pregenual and dorsal anterior cingulate cortex and elevated activity in the thalamus, caudate, anterior hippocampus and amygdala. Compared to the ALE meta analysis of PET/SPECT studies, a study using multi-kernel density analysis reported hyperactivity only in the pulvinar nuclei of the thalamus.
Brain Regions
Research on the brains of people with MDD usually shows disturbed patterns of interaction between multiple parts of the brain. Several areas of the brain are implicated in studies seeking to more fully understand the biology of depression:
Subgenual Cingulate
Studies have shown that Brodmann area 25, also known as subgenual cingulate, is metabolically overactive in treatment-resistant depression. This region is extremely rich in serotonin transporters and is considered as a governor for a vast network involving areas like hypothalamus and brain stem, which influences changes in appetite and sleep; the amygdala and insula, which affect the mood and anxiety; the hippocampus, which plays an important role in memory formation; and some parts of the frontal cortex responsible for self-esteem. Thus disturbances in this area or a smaller than normal size of this area contributes to depression. Deep brain stimulation has been targeted to this region in order to reduce its activity in people with treatment resistant depression.
Prefrontal Cortex
One review reported hypoactivity in the prefrontal cortex of those with depression compared to controls. The prefrontal cortex is involved in emotional processing and regulation, and dysfunction of this process may be involved in the aetiology of depression. One study on antidepressant treatment found an increase in PFC activity in response to administration of antidepressants. One meta analysis published in 2012 found that areas of the prefrontal cortex were hypoactive in response to negative stimuli in people with MDD. One study suggested that areas of the prefrontal cortex are part of a network of regions including dorsal and pregenual cingulate, bilateral middle frontal gyrus, insula and superior temporal gyrus that appear to be hypoactive in people with MDD. However the authors cautioned that the exclusion criteria, lack of consistency and small samples limit results.
Amygdala
The amygdala, a structure involved in emotional processing appears to be hyperactive in those with major depressive disorder. The amygdala in unmedicated depressed persons tended to be smaller than in those that were medicated, however aggregate data shows no difference between depressed and healthy persons. During emotional processing tasks right amygdala is more active than the left, however there is no differences during cognitive tasks, and at rest only the left amygdala appears to be more hyperactive. One study, however, found no difference in amygdala activity during emotional processing tasks.
Hippocampus
Atrophy of the hippocampus has been observed during depression, consistent with animal models of stress and neurogenesis.
Stress can cause depression and depression-like symptoms through monoaminergic changes in several key brain regions as well as suppression in hippocampal neurogenesis. This leads to alteration in emotion and cognition related brain regions as well as HPA axis dysfunction. Through the dysfunction, the effects of stress can be exacerbated including its effects on 5-HT. Furthermore, some of these effects are reversed by antidepressant action, which may act by increasing hippocampal neurogenesis. This leads to a restoration in HPA activity and stress reactivity, thus restoring the deleterious effects induced by stress on 5-HT.
The hypothalamic-pituitary-adrenal axis is a chain of endocrine structures that are activated during the body’s response to stressors of various sorts. The HPA axis involves three structure, the hypothalamus which release CRH that stimulates the pituitary gland to release ACTH which stimulates the adrenal glands to release cortisol. Cortisol has a negative feedback effect on the pituitary gland and hypothalamus. In people with MDD this often shows increased activation in depressed people, but the mechanism behind this is not yet known. Increased basal cortisol levels and abnormal response to dexamethasone challenges have been observed in people with MDD. Early life stress has been hypothesized as a potential cause of HPA dysfunction. HPA axis regulation may be examined through a dexamethasone suppression tests, which tests the feedback mechanisms. Non-suppression of dexamethasone is a common finding in depression, but is not consistent enough to be used as a diagnostic tool. HPA axis changes may be responsible for some of the changes such as decreased bone mineral density and increased weight found in people with MDD. One drug, ketoconazole, currently under development has shown promise in treating MDD.
Hippocampal Neurogenesis
Reduced hippocampal neurogenesis leads to a reduction in hippocampal volume. A genetically smaller hippocampus has been linked to a reduced ability to process psychological trauma and external stress, and subsequent predisposition to psychological illness. Depression without familial risk or childhood trauma has been linked to a normal hippocampal volume but localised dysfunction.
Animal Models
A number of animal models exist for depression, but they are limited in that depression involves primarily subjective emotional changes. However, some of these changes are reflected in physiology and behaviour, the latter of which is the target of many animal models. These models are generally assessed according to four facets of validity; the reflection of the core symptoms in the model; the predictive validity of the model; the validity of the model with regard to human characteristics of aetiology; and the biological plausibility.
Different models for inducing depressive behaviours have been utilised; neuroanatomical manipulations such as olfactory bulbectomy or circuit specific manipulations with optogenetics; genetic models such as 5-HT1A knockout or selectively bred animals; models involving environmental manipulation associated with depression in humans, including chronic mild stress, early life stress and learned helplessness. The validity of these models in producing depressive behaviours may be assessed with a number of behavioural tests. Anhedonia and motivational deficits may, for example, be assessed via examining an animal’s level of engagement with rewarding stimuli such as sucrose or intracranial self-stimulation. Anxious and irritable symptoms may be assessed with exploratory behaviour in the presence of a stressful or novelty environment, such as the open field test, novelty suppressed feeding, or the elevated plus-maze. Fatigue, psychomotor poverty, and agitation may be assessed with locomotor activity, grooming activity, and open field tests.
Animal models possess a number of limitations due to the nature of depression. Some core symptoms of depression, such as rumination, low self-esteem, guilt, and depressed mood cannot be assessed in animals as they require subjective reporting. From an evolutionary standpoint, the behaviour correlates of defeats of loss are thought to be an adaptive response to prevent further loss. Therefore, attempts to model depression that seeks to induce defeat or despair may actually reflect adaption and not disease. Furthermore, while depression and anxiety are frequently comorbid, dissociation of the two in animal models is difficult to achieve. Pharmacological assessment of validity is frequently disconnected from clinical pharmacotherapeutics in that most screening tests assess acute effects, while antidepressants normally take a few weeks to work in humans.
Neurocircuits
Regions involved in reward are common targets of manipulation in animal models of depression, including the nucleus accumbens (NAc), ventral tegmental area (VTA), ventral pallidum (VP), lateral habenula (LHb) and medial prefrontal cortex (mPFC). Tentative fMRI studies in humans demonstrate elevated LHb activity in depression. The lateral habenula projects to the RMTg to drive inhibition of dopamine neurons in the VTA during omission of reward. In animal models of depression, elevated activity has been reported in LHb neurons that project to the ventral tegmental area (ostensibly reducing dopamine release). The LHb also projects to aversion reactive mPFC neurons, which may provide an indirect mechanism for producing depressive behaviours. Learned helplessness induced potentiation of LHb synapses are reversed by antidepressant treatment, providing predictive validity. A number of inputs to the LHb have been implicated in producing depressive behaviours. Silencing GABAergic projections from the NAc to the LHb reduces conditioned place preference induced in social aggression, and activation of these terminals induces CPP. Ventral pallidum firing is also elevated by stress induced depression, an effect that is pharmacologically valid, and silencing of these neurons alleviates behavioural correlates of depression. Tentative in vivo evidence from people with MDD suggests abnormalities in dopamine signalling. This led to early studies investigating VTA activity and manipulations in animal models of depression. Massive destruction of VTA neurons enhances depressive behaviours, while VTA neurons reduce firing in response to chronic stress. However, more recent specific manipulations of the VTA produce varying results, with the specific animal model, duration of VTA manipulation, method of VTA manipulation, and subregion of VTA manipulation all potentially leading to differential outcomes. Stress and social defeat induced depressive symptoms, including anhedonia, are associated with potentiation of excitatory inputs to Dopamine D2 receptor-expressing medium spiny neurons (D2-MSNs) and depression of excitatory inputs to Dopamine D1 receptor-expressing medium spiny neurons (D1-MSNs). Optogenetic excitation of D1-MSNs alleviates depressive symptoms and is rewarding, while the same with D2-MSNs enhances depressive symptoms. Excitation of glutaminergic inputs from the ventral hippocampus reduces social interactions, and enhancing these projections produces susceptibility to stress-induced depression. Manipulations of different regions of the mPFC can produce and attenuate depressive behaviours. For example, inhibiting mPFC neurons specifically in the intralimbic cortex attenuates depressive behaviours. The conflicting findings associated with mPFC stimulation, when compared to the relatively specific findings in the infralimbic cortex, suggest that the prelimbic cortex and infralimbic cortex may mediate opposing effects. mPFC projections to the raphe nuclei are largely GABAergic and inhibit the firing of serotonergic neurons. Specific activation of these regions reduce immobility in the forced swim test but do not affect open field or forced swim behaviour. Inhibition of the raphe shifts the behavioural phenotype of uncontrolled stress to a phenotype closer to that of controlled stress.
Altered Neuroplasticity
Recent studies have called attention to the role of altered neuroplasticity in depression. A review found a convergence of three phenomena:
Chronic stress reduces synaptic and dendritic plasticity;
Depressed subjects show evidence of impaired neuroplasticity (e.g. shortening and reduced complexity of dendritic trees); and
Anti-depressant medications may enhance neuroplasticity at both a molecular and dendritic level.
The conclusion is that disrupted neuroplasticity is an underlying feature of depression, and is reversed by antidepressants.
Blood levels of BDNF in people with MDD increase significantly with antidepressant treatment and correlate with decrease in symptoms. Post mortem studies and rat models demonstrate decreased neuronal density in the prefrontal cortex thickness in people with MDD. Rat models demonstrate histological changes consistent with MRI findings in humans, however studies on neurogenesis in humans are limited. Antidepressants appear to reverse the changes in neurogenesis in both animal models and humans.
Inflammation
Various reviews have found that general inflammation may play a role in depression. One meta analysis of cytokines in people with MDD found increased levels of pro-inflammatory IL-6 and TNF-a levels relative to controls. The first theories came about when it was noticed that interferon therapy caused depression in a large number of people receiving it. Meta analysis on cytokine levels in people with MDD have demonstrated increased levels of IL-1, IL-6, C-reactive protein, but not IL-10. Increased numbers of T-Cells presenting activation markers, levels of neopterin, IFN gamma, sTNFR, and IL-2 receptors have been observed in depression. Various sources of inflammation in depressive illness have been hypothesized and include trauma, sleep problems, diet, smoking and obesity. Cytokines, by manipulating neurotransmitters, are involved in the generation of sickness behaviour, which shares some overlap with the symptoms of depression. Neurotransmitters hypothesized to be affected include dopamine and serotonin, which are common targets for antidepressant drugs. Induction of indolamine-2,3 dioxygenease by cytokines has been proposed as a mechanism by which immune dysfunction causes depression. One review found normalization of cytokine levels after successful treatment of depression. A meta analysis published in 2014 found the use of anti-inflammatory drugs such as NSAIDs and investigational cytokine inhibitors reduced depressive symptoms. Exercise can act as a stressor, decreasing the levels of IL-6 and TNF-a and increasing those of IL-10, an anti-inflammatory cytokine.
Inflammation is also intimately linked with metabolic processes in humans. For example, low levels of Vitamin D have been associated with greater risk for depression. The role of metabolic biomarkers in depression is an active research area. Recent work has explored the potential relationship between plasma sterols and depressive symptom severity.
Oxidative Stress
A marker of DNA oxidation, 8-Oxo-2′-deoxyguanosine, has been found to be increased in both the plasma and urine of people with MDD. This along with the finding of increased F2-isoprostanes levels found in blood, urine and cerebrospinal fluid indicate increased damage to lipids and DNA in people with MDD. Studies with 8-Oxo-2′ Deoxyguanosine varied by methods of measurement and type of depression, but F2-Isoprostane level was consistent across depression types. Authors suggested lifestyle factors, dysregulation of the HPA axis, immune system and autonomics nervous system as possible causes. Another meta-analysis found similar results with regards to oxidative damage products as well as decreased oxidative capacity. Oxidative DNA damage may play a role in MDD.
Mitochondrial Dysfunction:
Increased markers of oxidative stress relative to controls have been found in people with MDD. These markers include high levels of RNS and ROS which have been shown to influence chronic inflammation, damaging the electron transport chain and biochemical cascades in mitochondria. This lowers the activity of enzymes in the respiratory chain resulting in mitochondrial dysfunction. The brain is a highly energy-consuming and has little capacity to store glucose as glycogen and so depends greatly on mitochondria. Mitochondrial dysfunction has been linked to the dampened neuroplasticity observed in depressed brains.
Large-Scale Brain Network Theory
Instead of studying one brain region, studying large scale brain networks is another approach to understanding psychiatric and neurological disorders, supported by recent research that has shown that multiple brain regions are involved in these disorders. Understanding the disruptions in these networks may provide important insights into interventions for treating these disorders. Recent work suggests that at least three large-scale brain networks are important in psychopathology.
Central Executive Network
The central executive network is made up of fronto-parietal regions, including dorsolateral prefrontal cortex and lateral posterior parietal cortex. This network is involved in high level cognitive functions such as maintaining and using information in working memory, problem solving, and decision making. Deficiencies in this network are common in most major psychiatric and neurological disorders, including depression. Because this network is crucial for everyday life activities, those who are depressed can show impairment in basic activities like test taking and being decisive.
Default Mode Network
The default mode network includes hubs in the prefrontal cortex and posterior cingulate, with other prominent regions of the network in the medial temporal lobe and angular gyrus. The default mode network is usually active during mind-wandering and thinking about social situations. In contrast, during specific tasks probed in cognitive science (for example, simple attention tasks), the default network is often deactivated. Research has shown that regions in the default mode network (including medial prefrontal cortex and posterior cingulate) show greater activity when depressed participants ruminate (that is, when they engage in repetitive self-focused thinking) than when typical, healthy participants ruminate. People with MDD also show increased connectivity between the default mode network and the subgenual cingulate and the adjoining ventromedial prefrontal cortex in comparison to healthy individuals, individuals with dementia or with autism. Numerous studies suggest that the subgenual cingulate plays an important role in the dysfunction that characterizes major depression. The increased activation in the default mode network during rumination and the atypical connectivity between core default mode regions and the subgenual cingulate may underlie the tendency for depressed individual to get “stuck” in the negative, self-focused thoughts that often characterise depression. However, further research is needed to gain a precise understanding of how these network interactions map to specific symptoms of depression.
Salience Network
The salience network is a cingulate-frontal operculum network that includes core nodes in the anterior cingulate and anterior insula. A salience network is a large-scale brain network involved in detecting and orienting the most pertinent of the external stimuli and internal events being presented. Individuals who have a tendency to experience negative emotional states (scoring high on measures of neuroticism) show an increase in the right anterior insula during decision-making, even if the decision has already been made. This atypically high activity in the right anterior insula is thought to contribute to the experience of negative and worrisome feelings. In MDD, anxiety is often a part of the emotional state that characterises depression.
Lamotrigine, sold as the brand name Lamictal among others, is an anticonvulsant medication used to treat epilepsy and to delay or prevent the recurrence of depressive episodes in bipolar disorder. For epilepsy, this includes focal seizures, tonic-clonic seizures, and seizures in Lennox-Gastaut syndrome. In bipolar disorder, lamotrigine has not been shown to reliably treat acute depression; but for patients with bipolar disorder who are not currently symptomatic, it appears to be effective in reducing the risk of future episodes of depression.
Common side effects include nausea, sleepiness, headache, vomiting, trouble with coordination, and rash. Serious side effects include lack of red blood cells, increased risk of suicide, Stevens-Johnson syndrome, and allergic reactions. Concerns exist that use during pregnancy or breastfeeding may result in harm. Lamotrigine is a phenyltriazine, making it chemically different from other anticonvulsants. Its mechanism of action is not clear, but it appears to inhibit release of excitatory neurotransmitters via voltage-sensitive sodium channels in neurons.
Lamotrigine was first marketed in the United Kingdom in 1991, and approved for use in the United States in 1994. It is on the World Health Organization’s List of Essential Medicines. In 2019, it was the 71st most commonly prescribed medication in the United States, with more than 10 million prescriptions.
Brief History
1991 – Lamotrigine is first used in the United Kingdom as an anticonvulsant medication.
December 1994 – Lamotrigine was first approved for use in the United States and, that for the treatment of partial seizures.
August 1998 – For use as adjunctive treatment of Lennox-Gastaut syndrome in paediatric and adult patients, new dosage form: chewable dispersible tablets.
December 1998 – For use as monotherapy for treatment of partial seizures in adult patients when converting from a single enzyme-inducing anticonvulsant drug.
January 2003 – For use as adjunctive therapy for partial seizures in paediatric patients as young as two years of age.
June 2003 – Approved for maintenance treatment of Bipolar II disorder; the first such medication since lithium.
January 2004 – For use as monotherapy for treatment of partial seizures in adult patients when converting from the anti-epileptic drug valproate (including valproic acid).
Medical Uses
Epilepsy
Lamotrigine is considered a first-line drug for primary generalized tonic-clonic seizures (includes simple partial, complex partial, and secondarily generalized seizures such as focal-onset tonic-clonic seizures). It is also used as an alternative or adjuvant medication for partial seizures, such as absence seizure, myoclonic seizure, and atonic seizures. A 2020 review on the use of Lamotrigine as an add-on therapy for drug resistant generalized tonic-clonic seizures was unable to come to conclusions to inform clinical practice. Although low-certainty evidence suggest that it reduces generalised tonic-clonic seizures by 50% the level of uncertainty indicates that the actual findings could be significantly different. Another 2020 Cochrane review examining the use of lamotrigine as an add-on therapy for drug-resistant focal epilepsy found it to be effective for reducing seizure frequency and was well tolerated.
Lennox-Gastaut Syndrome
Lamotrigine is one of a small number of FDA-approved therapies for the form of epilepsy known as Lennox-Gastaut syndrome. It reduces the frequency of LGS seizures, and is one of two medications known to decrease the severity of drop attacks. Combination with valproate is common, but this increases the risk of lamotrigine-induced rash, and necessitates reduced dosing due to the interaction of these drugs.
Bipolar Disorder
Lamotrigine is approved in the US for maintenance treatment of bipolar I disorder and bipolar II disorder. While the anticonvulsants carbamazepine and valproate are predominantly antimanics, lamotrigine has demonstrated efficacy only in preventing or reducing the risk of recurrent depressive episodes of bipolar disorder. The drug seems ineffective in the treatment of current rapid-cycling, acute mania, or acute depression in bipolar disorder.
Lamotrigine has not demonstrated clear efficacy in treating acute mood episodes, either mania or depression. It has not demonstrated effectiveness in treating acute mania, and there is controversy regarding the drug’s effectiveness in treating acute bipolar depression. A paper written in 2008 by Nassir et al. reviewed evidence from trials that were unpublished and not referenced in the 2002 APA guidelines, and it concludes that lamotrigine has “very limited, if any, efficacy in the treatment of acute bipolar depression”. A 2008 paper by Calabrese et al. examined much of the same data, and found that in five placebo-controlled studies, lamotrigine did not significantly differ from placebo in the treatment of bipolar depression. However, in a meta-analysis of these studies conducted in 2008, Geddes, Calabrese and Goodwin found that lamotrigine was effective in individuals with bipolar depression, with a number needed to treat (NNT) of 11, or 7 in severe depression.
A 2013 review about lamotrigine concluded that it is recommended in bipolar maintenance when depression is prominent and that more research is needed in regard to its role in the treatment of acute bipolar depression and unipolar depression. No information to recommend its use in other psychiatric disorders was found.
Schizophrenia
Lamotrigine, as a monotherapy, is not substantially effective against schizophrenia. However; various publications and textbooks have expressed that lamotrigine could be added to clozapine as augmentation therapy against partial or non-responding schizophrenic patients. Patients had statistically significant improvements in positive, negative and affective symptoms. Lamotrigine does not have a statistically significant effect with antipsychotics other than clozapine, such as: olanzapine, risperidone, haloperidol, zuclopenthixol, etc.
Other Uses
Off-label uses include the treatment of peripheral neuropathy, trigeminal neuralgia, cluster headaches, migraines, visual snow, and reducing neuropathic pain, although a systematic review conducted in 2013 concluded that well-designed clinical trials have shown no benefit for lamotrigine in neuropathic pain. Off-label psychiatric usage includes the treatment of treatment-resistant obsessive-compulsive disorder, depersonalisation disorder, hallucinogen persisting perception disorder, schizoaffective disorder, and borderline personality disorder.
It has not been shown to be useful in post-traumatic stress disorder.
Side Effects
Lamotrigine prescribing information has a black box warning about life-threatening skin reactions, including Stevens-Johnson syndrome (SJS), DRESS syndrome, and toxic epidermal necrolysis (TEN). The manufacturer states that nearly all cases appear in the first two to eight weeks of therapy. Patients should seek medical attention for any unexpected skin rash, as its presence is an indication of a possible serious or even deadly side effect of the drug. Not all rashes that occur while taking lamotrigine progress to SJS or TEN. Between 5 and 10% of patients will develop a rash, but only one in a thousand patients will develop a serious rash. Rash and other skin reactions are more common in children, so this medication is often reserved for adults. For patients whose lamotrigine has been stopped after development of a rash, rechallenge with lamotrigine is also a viable option. However, it is not applicable for very serious cases. The incidence of these eruptions increases in patients who are currently on, or recently discontinued a valproate-type anticonvulsant drug, as these medications interact in such a way that the clearance of both is decreased and the effective dose of lamotrigine is increased.
Side effects such as rash, fever, and fatigue are very serious, as they may indicate incipient SJS, TEN, DRESS syndrome, or aseptic meningitis. Other side effects include loss of balance or coordination, double vision, crossed eyes, pupil constriction, blurred vision, dizziness and lack of coordination, drowsiness, insomnia, anxiety, vivid dreams or nightmares, dry mouth, mouth ulcers, memory problems, mood changes, itchiness, runny nose, cough, nausea, indigestion, abdominal pain, weight loss, missed or painful menstrual periods, and vaginitis. The side-effects profile varies for different patient populations. Overall adverse effects in treatment are similar between men, women, geriatric, paediatric and racial groups.
Lamotrigine has been associated with a decrease in white blood cell count (leukopenia). Lamotrigine does not prolong QT/QTc in TQT studies in healthy subjects.
In people taking antipsychotics, cases of lamotrigine-precipitated neuroleptic malignant syndrome have been reported.
In 2018, the FDA required a new warning for the risk of hemophagocytic lymphohistiocytosis. This reaction can occur between days to weeks after starting the treatment.
Women
Women are more likely than men to have side effects. This is the opposite of most other anticonvulsants.
Some evidence shows interactions between lamotrigine and female hormones, which can be of particular concern for women on oestrogen-containing hormonal contraceptives. Ethinylestradiol, an ingredient of such contraceptives, has been shown to decrease serum levels of lamotrigine. Women starting an oestrogen-containing oral contraceptive may need to increase the dosage of lamotrigine to maintain its level of efficacy. Likewise, women may experience an increase in lamotrigine side effects upon discontinuation of birth control pills. This may include the “pill-free” week where lamotrigine serum levels have been shown to increase twofold.
Pregnancy and Breastfeeding
Many studies have found no association between lamotrigine exposure in utero and birth defects, while those that have found an association have found only slight associations with minor malformations such as cleft palates. Review studies have found that overall rates of congenital malformations in infants exposed to lamotrigine in utero are relatively low (1-4%), which is similar to the rate of malformations in the general population. It is known that lamotrigine is a weak inhibitor of human dihydrofolate reductase (DHFR) and other, more powerful, human DHFR inhibitors such as methotrexate are known to be teratogenic.
Lamotrigine is expressed in breast milk; the manufacturer does not recommend breastfeeding during treatment. However, recent studies suggest that lamotrigine is safe to use while breastfeeding. A frequently updated review of scientific literature rates lamotrigine as L3: moderately safe.
Other Types of Effects
Lamotrigine binds to melanin-containing tissues such as the iris of the eye or melanin-rich skin. The long-term consequences of this are unknown.
GlaxoSmithKline investigated lamotrigine for the treatment of ADHD with inconclusive results. No detrimental effects on cognitive function were observed; however, the only statistical improvement in core ADHD symptoms was an improvement on a Paced Auditory Serial Addition Test (PASAT) that measures auditory processing speed and calculation ability. Another study reported that lamotrigine might be a safe and effective treatment option for adult ADHD comorbid with bipolar and recurrent depression.
Lamotrigine is known to affect sleep. Studies with small numbers of patients (10-15) reported that lamotrigine increases the duration of REM sleep, decreases the number of phase shifts, and decreases the duration of slow-wave sleep, and that there was no effect on vigilance, daytime somnolence and cognitive function. However, a retrospective study of 109 patients’ medical records found that 6.7% of patients experienced an “alerting effect” resulting in intolerable insomnia, for which the treatment had to be discontinued.
Lamotrigine can induce a type of seizure known as a myoclonic jerk, which tends to happen soon after the use of the medication. When used in the treatment of myoclonic epilepsies such as juvenile myoclonic epilepsy, lower doses (and lower plasma levels) are usually needed, as even moderate doses of this drug can induce seizures, including tonic-clonic seizures, which can develop into status epilepticus, which is a medical emergency. It can also cause myoclonic status epilepticus.
In overdose, lamotrigine can cause uncontrolled seizures in most people. Reported results in overdoses involving up to 15 g include increased seizures, coma, and death.
Pharmacology
Mechanism of Action
Lamotrigine is a member of the sodium channel blocking class of antiepileptic drugs. This may suppress the release of glutamate and aspartate, two dominant excitatory neurotransmitters in the central nervous system. It is generally accepted to be a member of the sodium channel blocking class of antiepileptic drugs, but it could have additional actions, since it has a broader spectrum of action than other sodium channel antiepileptic drugs such as phenytoin and is effective in the treatment of the depressed phase of bipolar disorder, whereas other sodium channel-blocking antiepileptic drugs are not, possibly on account of its sigma receptor activity. In addition, lamotrigine shares few side effects with other, unrelated anticonvulsants known to inhibit sodium channels, which further emphasizes its unique properties.
It is a triazine derivate that inhibits voltage-sensitive sodium channels, leading to stabilisation of neuronal membranes. It also blocks L-, N-, and P-type calcium channels and weakly inhibits the serotonin 5-HT3 receptor. These actions are thought to inhibit release of glutamate at cortical projections in the ventral striatum limbic areas, and its neuroprotective and anti-glutamatergic effects have been pointed out as promising contributors to its mood stabilising activity. Observations that lamotrigine reduced γ-aminobutyric acid (GABA) A receptor-mediated neurotransmission in rat amygdala, suggest that a GABAergic mechanism may also be involved. It appears that lamotrigine does not increase GABA blood levels in humans.
Lamotrigine does not have pronounced effects on any of the usual neurotransmitter receptors that anticonvulsants affect (adrenergic, dopamine D1 and D2, muscarinic, GABA, histaminergic H1, serotonin 5-HT2, and N-methyl-D-aspartate). Inhibitory effects on 5-HT, norepinephrine, and dopamine transporters are weak. Lamotrigine is a weak inhibitor of dihydrofolate reductase, but whether this effect is sufficient to contribute to a mechanism of action or increases risk to the foetus during pregnancy is not known. Early studies of lamotrigine’s mechanism of action examined its effects on the release of endogenous amino acids from rat cerebral cortex slices in vitro. As is the case for antiepileptic drugs that act on voltage-dependent sodium channels, lamotrigine thereby inhibits the release of glutamate and aspartate, which is evoked by the sodium-channel activator veratrine, and was less effective in the inhibition of acetylcholine or GABA release. At high concentrations, it had no effect on spontaneous or potassium-evoked amino acid release.
These studies suggested that lamotrigine acts presynaptically on voltage-gated sodium channels to decrease glutamate release. Several electrophysiological studies have investigated the effects of lamotrigine on voltage-gated sodium channels. For example, lamotrigine blocked sustained repetitive firing in cultured mouse spinal cord neurons in a concentration-dependent manner, at concentrations that are therapeutically relevant in the treatment of human seizures. In cultured hippocampal neurons, lamotrigine reduced sodium currents in a voltage-dependent manner, and at depolarised potentials showed a small frequency-dependent inhibition. These and a variety of other results indicate that the antiepileptic effect of lamotrigine, like those of phenytoin and carbamazepine, is at least in part due to use- and voltage-dependent modulation of fast voltage-dependent sodium currents. However, lamotrigine has a broader clinical spectrum of activity than phenytoin and carbamazepine and is recognised to be protective against generalised absence epilepsy and other generalised epilepsy syndromes, including primary generalised tonic-clonic seizures, juvenile myoclonic epilepsy, and Lennox-Gastaut syndrome.
The basis for this broader spectrum of activity of lamotrigine is unknown, but could relate to actions of the drug on voltage-gated calcium channels. Lamotrigine blocks T-type calcium channels weakly, if at all. However, it does inhibit native and recombinant high voltage–gated calcium channels (N- and P/Q/R-types) at therapeutic concentrations. Whether this activity on calcium channels accounts for lamotrigine’s broader clinical spectrum of activity in comparison with phenytoin and carbamazepine remains to be determined.
It antagonises these receptors with the following IC50 values:
5-HT3, IC50 = 18 μM
σ receptors, IC50 = 145 μM
Pharmacokinetics
The pharmacokinetics of lamotrigine follow first-order kinetics, with a half-life of 29 hours and volume of distribution of 1.36 L/kg. Lamotrigine is rapidly and completely absorbed after oral administration. Its absolute bioavailability is 98% and its plasma Cmax occurs from 1.4 to 4.8 hours. Available data indicate that its bioavailability is not affected by food. Estimate of the mean apparent volume of distribution of lamotrigine following oral administration ranges from 0.9 to 1.3 L/kg. This is independent of dose and is similar following single and multiple doses in both patients with epilepsy and in healthy volunteers.
Lamotrigine is inactivated by glucuronidation in the liver. Lamotrigine is metabolised predominantly by glucuronic acid conjugation. Its major metabolite is an inactive 2-n-glucuronide conjugate.
Lamotrigine has fewer drug interactions than many anticonvulsant drugs, although pharmacokinetic interactions with carbamazepine, phenytoin and other hepatic enzyme inducing medications may shorten half-life. Dose adjustments should be made on clinical response, but monitoring may be of benefit in assessing compliance.
The capacity of available tests to detect potentially adverse consequences of melanin binding is unknown. Clinical trials excluded subtle effects and optimal duration of treatment. There are no specific recommendations for periodic ophthalmological monitoring. Lamotrigine binds to the eye and melanin-containing tissues which can accumulate over time and may cause toxicity. Prescribers should be aware of the possibility of long-term ophthalmologic effects and base treatment on clinical response. Patient compliance should be periodically reassessed with lab and medical testing of liver and kidney function to monitor progress or side effects.
Society and Culture
Brand Names
Lamotrigine was originally brought to market by GlaxoSmithKline, trademarked as Lamictal; it is also available in generic form under many brand names worldwide.
Cyclothymia, also known as cyclothymic disorder, is a mental and behavioural disorder that involves numerous periods of symptoms of depression and periods of symptoms of elevated mood.
These symptoms, however, are not sufficient to be a major depressive episode or a hypomanic episode. Symptoms must last for more than one year in children and two years in adults.
The cause of cyclothymia is unknown. Risk factors include a family history of bipolar disorder. Cyclothymia differs from bipolar in that major depression, mania, or hypomania have never occurred.
Treatment is generally with counselling and mood stabilisers such as lithium. It is estimated that 0.4-1% of people have cyclothymia at some point in their life. Onset is typically in late childhood to early adulthood. Males and females are affected equally often.
Brief History
In 1883, Karl Ludwig Kahlbaum identified a disorder characterised by recurring mood cycles. The disorder contained both melancholic and manic episodes that occurred in a milder form than in bipolar disorder. This condition was coined “cyclothymia” by Kahlbaum and his student Ewald Hecker. Kahlbaum developed his theory of cyclothymia through his work with people presenting with these symptoms at the Kahlbaum Sanitarium in Goerlitz, Silesia (Germany). He was recognised as a leading hypnotherapist and psychotherapist of his day. He was a progressive in the field of mental health, believing that mental illness should not carry a stigma and that people dealing with mental health issues should be treated humanely. Kalhbaum was the first to recognise that people with cyclothymia often do not seek help for the disorder due to its mild symptoms.
Cyclothymia has been conceptualised in a variety of ways, including as a subtype of bipolar disorder, a temperament, a personality trait, and a personality disorder. There is also an argument that cyclothymia should be considered a neurodevelopmental disorder. The two defining features of the disorder, according to DSM-5, are the presence of depressive and hypomanic symptoms, not meeting the threshold for a depressive or hypomanic episode. Cyclothymia is also classified as a subtype of bipolar disorder in DSM-5, but some researchers disagree with this classification and argue that it should be primarily defined as an exaggeration of mood and emotional instability. In the past, cyclothymia has been conceptualised to include other characteristics in addition to the flux between depression and hypomania, such as mood reactivity, impulsivity, and anxiety.
Symptoms
People with cyclothymia experience both depressive phases and hypomanic phases (which are less severe than a full hypomanic episode). The depressive and manic symptoms in cyclothymia last for variable amounts of time due to the unstable and reactive nature of the disorder. The depressive phases are similar to major depressive disorder and are characterised by dulled thoughts and sensations and the lack of motivation for intellectual or social activities. Most people with cyclothymia are generally fatigued and tend to sleep frequently and for long periods of time. However, other people experience insomnia.
Other symptoms of cyclothymic depression include indifference toward people or activities that used to be extremely important. Cyclothymic depression also leads to difficulty making decisions. In addition, people with this condition tend to be critical and complain easily. Suicidal thoughts are common, even in mild forms of cyclothymia. In the depressive state, people with cyclothymia also experience physical complaints including frequent headaches, tightness in the head and chest, an empty sensation in the head, weakness, weight loss, and hair loss.
The distinguishing factor between typical depression and cyclothymic depression is that in cyclothymic depression, there are instances of hypomania. People with cyclothymia can switch from the depressive state to the hypomanic state without warning to them or others. The duration and frequency of phases is unpredictable.
In the hypomanic state, people’s thoughts become faster and they become more sociable and talkative. They may engage in spending sprees, spontaneous actions, have heightened self-esteem, and greater vanity. In contrast to a regular manic state that would be associated with bipolar I, symptoms in the hypomanic phase generally occur in a less severe form.
Comorbidities
Cyclothymia commonly occurs in conjunction with other disorders. Between 20-50 percent of people with depression, anxiety, and related disorders also have cyclothymia. When people with cyclothymia seek mental health resources it tends to be for symptoms of their comorbid condition rather than for their symptoms of cyclothymia. In children and adolescents, the most common comorbidities with cyclothymia are anxiety disorders, impulse control issues, eating disorders, and ADHD. In adults, cyclothymia also tends to be comorbid with impulse control issues. Sensation-seeking behaviours occur in hypomanic states. These often include gambling and compulsive sexuality in men, or compulsive buying and binge eating in women.
In addition to sensation-related disorders, cyclothymia has also been associated with atypical depression. In one study, a connection was found between interpersonal sensitivity, mood reactivity (i.e. responding to actual or potential positive events with brighter mood), and cyclothymic mood swings, all of which are symptoms of atypical depression. Cyclothymia also tends to occur in conjunction with separation anxiety, where a person has anxiety as a result of separation from a caregiver, friend, or loved one. Other issues that tend to co-occur with cyclothymia include social anxiety, fear of rejection and a tendency toward hostility to those connected with past pain and rejection. People with cyclothymia tend to seek intense interpersonal relationships when in a hypomanic state and isolation when in a depressed state. This generally leads to short, tumultuous relationships.
Causes
The cause is unknown. Risk factors include a family history of bipolar disorder.
First-degree relatives of people with cyclothymia have major depressive disorder, bipolar I disorder, and bipolar II disorder more often than the general population. Substance-related disorders also may be at a higher risk within the family. First-degree relatives of a bipolar I individuals may have a higher risk of cyclothymic disorder than the general population.
Diagnosis
Cyclothymia is classified in DSM-5 as a subtype of bipolar disorder. The criteria are:
Periods of elevated mood and depressive symptoms for at least half the time during the last two years for adults and one year for children and teenagers.
Periods of stable moods last only two months at most.
Symptoms create significant problems in one or more areas of life.
Symptoms do not meet the criteria for bipolar disorder, major depression, or another mental disorder.
Symptoms are not caused by substance use or a medical condition.
The DSM-5 criteria for cyclothymia are restrictive according to some researchers. This affects the diagnosis of cyclothymia because fewer people get diagnosed than potentially could. This means that a person who has some symptoms of the disorder might not be able to get treatment because they do not meet all of the necessary criteria described in DSM-5. Furthermore, it also leads to more attention being placed on depression and other bipolar-spectrum disorders because if a person does not meet all the criteria for cyclothymia they are often given a depression or bipolar spectrum diagnosis. Improper diagnosis may lead some people with cyclothymia to be treated for a comorbid disorder rather than having their cyclothymic tendencies addressed.
Cyclothymia is often not recognised by the affected individual or medical professionals due to its ostensibly mild symptoms. In addition, it is difficult to identify and classify. Due to disagreement and misconceptions among health and mental health professionals, cyclothymia is often diagnosed as “bipolar not otherwise specified”. Cyclothymia is also often confused with borderline personality disorder due to their similar symptoms, especially in older adolescents and young adults.
Most people with the disorder present in a depressive state, not realising that their hypomanic states are abnormal. Mild manic episodes tend to be interpreted as part of the person’s personality or simply a heightened mood. In addition, the disorder often manifests during childhood or adolescence, making it even more difficult for the person to distinguish between symptoms of the disorder and their personality. For example, people may think that they just suffer from mood swings and not realise that these are a result of a psychiatric condition.
Medication can be used in addition to behavioural approaches. However, mood stabilisers should be used before antidepressants, and if antidepressants are used they should be used with caution. Antidepressants are a concern due to the possibility of inducing hypomanic switches or rapid cycling.
Epidemiology
Cyclothymia, known today as cyclothymic disorder, tends to be underdiagnosed due to its low intensity. The exact rates for cyclothymia have not been widely studied. Some studies estimate that between 5 and 8% are affected at some point in their life whereas other studies suggest a rate ranging from 0.4 to 2.5%.
Males appear to be affected equally often, though women are more likely to receive treatment. Cyclothymia is diagnosed in around fifty percent of people with depression who are evaluated in psychiatric outpatient settings.
Etymology
Cyclothymia is derived from the Greek word κυκλοθυμία (from κῦκλος kyklos, “circle” and θυμός thymos, “mood, emotion”). Therefore, it means “to cycle or circle between moods or emotions”.
Research
Whether subtypes of bipolar disorder, such as cyclothymia, truly represent separate disorders or are part of a unique bipolar spectrum is debated in research. Cyclothymia is typically not described in research studies or diagnosed in clinical settings, making it less recognisable and less understood by professionals. This absence of cyclothymia in research and clinical settings suggests that cyclothymia is either being diagnosed as another mood disorder or as a non-affective psychiatric disorder or not coming to scientific or clinical attention due to a lack of diagnostic clarity or because the nature of cyclothymia is still highly contested. Additionally, the current diagnostic criterion for cyclothymia emphasizes that symptoms are persistent, which suggests that they are enduring traits rather than a psychological state, thus, it has been argued that it should be diagnosed as a personality disorder. Since the symptoms tend to overlap with personality disorders, the validity and distinction between these two diagnostic categories has been debated.
Lastly, the tendency of cyclothymia to be comorbid with other mental disorders makes diagnosis difficult. These issues prevent consensus on the definition of cyclothymia and its relationship with other mental disorders among researchers and clinicians. This lack of consensus on an operational definition and symptom presentation is especially pronounced with children and adolescents because the diagnostic criteria have not been adequately adapted to take into account their developmental level.
Society and Culture
Actor Stephen Fry has spoken about his experience with cyclothymia, which was depicted in the documentary Stephen Fry: The Secret Life of the Manic Depressive.
Singer Charlene Soraia had cyclothymia and wrote a song about her experiences with the disorder.
Dysthymia, also known as persistent depressive disorder (PDD), is a mental and behavioural disorder, specifically a disorder primarily of mood, consisting of the same cognitive and physical problems as depression, but with longer-lasting symptoms.
The concept was coined by Robert Spitzer as a replacement for the term “depressive personality” in the late 1970s.
As dysthymia is a chronic disorder, sufferers may experience symptoms for many years before it is diagnosed, if diagnosis occurs at all. As a result, they may believe that depression is a part of their character, so they may not even discuss their symptoms with doctors, family members or friends. In the DSM-5, dysthymia is replaced by persistent depressive disorder. This new condition includes both chronic major depressive disorder and the previous dysthymic disorder. The reason for this change is that there was no evidence for meaningful differences between these two conditions.
Epidemiology
Globally dysthymia occurs in about 105 million people a year (1.5% of the population). It is 38% more common in women (1.8% of women) than in men (1.3% of men). The lifetime prevalence rate of dysthymia in community settings appears to range from 3 to 6% in the United States. However, in primary care settings the rate is higher ranging from 5 to 15 percent. United States prevalence rates tend to be somewhat higher than rates in other countries.
Signs and Symptoms
Dysthymia characteristics include an extended period of depressed mood combined with at least two other symptoms which may include insomnia or hypersomnia, fatigue or low energy, eating changes (more or less), low self-esteem, or feelings of hopelessness. Poor concentration or difficulty making decisions are treated as another possible symptom. Irritability is one of the more common symptoms in children and adolescents.
Mild degrees of dysthymia may result in people withdrawing from stress and avoiding opportunities for failure. In more severe cases of dysthymia, people may withdraw from daily activities. They will usually find little pleasure in usual activities and pastimes.
Diagnosis of dysthymia can be difficult because of the subtle nature of the symptoms and patients can often hide them in social situations, making it challenging for others to detect symptoms. Additionally, dysthymia often occurs at the same time as other psychological disorders, which adds a level of complexity in determining the presence of dysthymia, particularly because there is often an overlap in the symptoms of disorders.
There is a high incidence of comorbid illness in those with dysthymia. Suicidal behaviour is also a particular problem with those with dysthymia. It is vital to look for signs of major depression, panic disorder, generalised anxiety disorder, alcohol and substance use disorders, and personality disorder.
Causes
There are no known biological causes that apply consistently to all cases of dysthymia, which suggests diverse origin of the disorder. However, there are some indications that there is a genetic predisposition to dysthymia: “The rate of depression in the families of people with dysthymia is as high as fifty percent for the early-onset form of the disorder”. Other factors linked with dysthymia include stress, social isolation, and lack of social support.
In a study using identical and fraternal twins, results indicated that there is a stronger likelihood of identical twins both having depression than fraternal twins. This provides support for the idea that dysthymia is in part caused by heredity.
Co-Occurring Conditions
Dysthymia often co-occurs with other mental disorders. A “double depression” is the occurrence of episodes of major depression in addition to dysthymia. Switching between periods of dysthymic moods and periods of hypomanic moods is indicative of cyclothymia, which is a mild variant of bipolar disorder.
“At least three-quarters of patients with dysthymia also have a chronic physical illness or another psychiatric disorder such as one of the anxiety disorders, cyclothymia, drug addiction, or alcoholism”. Common co-occurring conditions include major depression (up to 75%), anxiety disorders (up to 50%), personality disorders (up to 40%), somatoform disorders (up to 45%) and substance use disorders (up to 50%). People with dysthymia have a higher-than-average chance of developing major depression. A 10-year follow-up study found that 95% of dysthymia patients had an episode of major depression. When an intense episode of depression occurs on top of dysthymia, the state is called “double depression.”
Double Depression
Double depression occurs when a person experiences a major depressive episode on top of the already-existing condition of dysthymia. It is difficult to treat, as sufferers accept these major depressive symptoms as a natural part of their personality or as a part of their life that is outside of their control. The fact that people with dysthymia may accept these worsening symptoms as inevitable can delay treatment. When and if such people seek out treatment, the treatment may not be very effective if only the symptoms of the major depression are addressed, but not the dysthymic symptoms. Patients with double depression tend to report significantly higher levels of hopelessness than is normal. This can be a useful symptom for mental health services providers to focus on when working with patients to treat the condition. Additionally, cognitive therapies can be effective for working with people with double depression in order to help change negative thinking patterns and give individuals a new way of seeing themselves and their environment.
It has been suggested that the best way to prevent double depression is by treating the dysthymia. A combination of antidepressants and cognitive therapies can be helpful in preventing major depressive symptoms from occurring. Additionally, exercise and good sleep hygiene (e.g. improving sleep patterns) are thought to have an additive effect on treating dysthymic symptoms and preventing them from worsening.
Pathophysiology
There is evidence that there may be neurological indicators of early onset dysthymia. There are several brain structures (corpus callosum and frontal lobe) that are different in women with dysthymia than in those without dysthymia. This may indicate that there is a developmental difference between these two groups.
Another study, which used fMRI techniques to assess the differences between individuals with dysthymia and other people, found additional support for neurological indicators of the disorder. This study found several areas of the brain that function differently. The amygdala (associated with processing emotions such as fear) was more activated in dysthymia patients. The study also observed increased activity in the insula (which is associated with sad emotions). Finally, there was increased activity in the cingulate gyrus (which serves as the bridge between attention and emotion).
A study comparing healthy individuals to people with dysthymia indicates there are other biological indicators of the disorder. An anticipated result appeared as healthy individuals expected fewer negative adjectives to apply to them, whereas people with dysthymia expected fewer positive adjectives to apply to them in the future. Biologically these groups are also differentiated in that healthy individuals showed greater neurological anticipation for all types of events (positive, neutral, or negative) than those with dysthymia. This provides neurological evidence of the dulling of emotion that individuals with dysthymia have learned to use to protect themselves from overly strong negative feelings, compared to healthy people.
There is some evidence of a genetic basis for all types of depression, including dysthymia. A study using identical and fraternal twins indicated that there is a stronger likelihood of identical twins both having depression than fraternal twins. This provides support for the idea that dysthymia is caused in part by heredity.
A new model has recently surfaced in the literature regarding the HPA axis (structures in the brain that get activated in response to stress) and its involvement with dysthymia (e.g. phenotypic variations of corticotropin releasing hormone (CRH) and arginine vasopressin (AVP), and down-regulation of adrenal functioning) as well as forebrain serotonergic mechanisms. Since this model is highly provisional, further research is still needed.
Diagnosis
The Diagnostic and Statistical Manual of Mental Disorders IV (DSM-IV), published by the American Psychiatric Association, characterises dysthymic disorder. The essential symptom involves the individual feeling depressed for the majority of days, and parts of the day, for at least two years. Low energy, disturbances in sleep or in appetite, and low self-esteem typically contribute to the clinical picture as well. Sufferers have often experienced dysthymia for many years before it is diagnosed. People around them often describe the sufferer in words similar to “just a moody person”. Note the following diagnostic criteria:
During a majority of days for two years or more, the adult patient reports depressed mood, or appears depressed to others for most of the day.
When depressed, the patient has two or more of:
decreased or increased appetite
decreased or increased sleep (insomnia or hypersomnia)
Fatigue or low energy
Reduced self-esteem
Decreased concentration or problems making decisions
Feelings of hopelessness or pessimism
During this two-year period, the above symptoms are never absent longer than two consecutive months.
During the duration of the two-year period, the patient may have had a perpetual major depressive episode.
The patient has not had any manic, hypomanic, or mixed episodes.
The patient has never fulfilled criteria for cyclothymic disorder.
The symptoms are often not directly caused by a medical illness or by substances, including substance use or other medications.
The symptoms may cause significant problems or distress in social, work, academic, or other major areas of life functioning.
In children and adolescents, mood can be irritable, and duration must be at least one year, in contrast to two years needed for diagnosis in adults.
Early onset (diagnosis before age 21) is associated with more frequent relapses, psychiatric hospitalisations, and more co-occurring conditions. For younger adults with dysthymia, there is a higher co-occurrence in personality abnormalities and the symptoms are likely chronic. However, in older adults suffering from dysthymia, the psychological symptoms are associated with medical conditions and/or stressful life events and losses.
Dysthymia can be contrasted with major depressive disorder by assessing the acute nature of the symptoms. Dysthymia is far more chronic (long lasting) than major depressive disorder, in which symptoms may be present for as little as 2 weeks. Also Dysthymia often presents itself at an earlier age than Major Depressive Disorder.
Prevention
Though there is no clear-cut way to prevent dysthymia from occurring, some suggestions have been made. Since dysthymia will often first occur in childhood, it is important to identify children who may be at risk. It may be beneficial to work with children in helping to control their stress, increase resilience, boost self-esteem, and provide strong networks of social support. These tactics may be helpful in warding off or delaying dysthymic symptoms.
Treatment
Persistent depressive disorder can be treated with psychotherapy and pharmacotherapy. The overall rate and degree of treatment success is somewhat lower than for non-chronic depression, and a combination of psychotherapy and pharmacotherapy shows best results.
Therapy
Psychotherapy can be effective in treating dysthymia. In a meta-analytic study from 2010, psychotherapy had a small but significant effect when compared to control groups. However, psychotherapy is significantly less effective than pharmacotherapy in direct comparisons.
There are many different types of therapy, and some are more effective than others.
The empirically most studied type of treatment is cognitive-behavioural therapy.
This type of therapy is very effective for non-chronic depression, and it appears to be also effective for chronic depression.
Cognitive behavioural analysis system of psychotherapy (CBASP) has been designed specifically to treat PDD.
Empirical results on this form of therapy are inconclusive: While one study showed remarkably high treatment success rates, a later, even larger study showed no significant benefit of adding CBASP to treatment with antidepressants.
Schema therapy and psychodynamic psychotherapy have been used for PDD, though good empirical results are lacking.
Interpersonal psychotherapy has also been said to be effective in treating the disorder, though it only shows marginal benefit when added to treatment with antidepressants.
Medications
In a 2010 meta-analysis, the benefit of pharmacotherapy was limited to selective serotonin reuptake inhibitors (SSRIs) rather than tricyclic antidepressants (TCA).
According to a 2014 meta-analysis, antidepressants are at least as effective for persistent depressive disorder as for major depressive disorder. The first line of pharmacotherapy is usually SSRIs due to their purported more tolerable nature and reduced side effects compared to the irreversible monoamine oxidase inhibitors or tricyclic antidepressants. Studies have found that the mean response to antidepressant medications for people with dysthymia is 55%, compared with a 31% response rate to a placebo. The most commonly prescribed antidepressants/SSRIs for dysthymia are escitalopram, citalopram, sertraline, fluoxetine, paroxetine, and fluvoxamine. It often takes an average of 6-8 weeks before the patient begins to feel these medications’ therapeutic effects. Additionally, STAR*D, a multi-clinic governmental study, found that people with overall depression will generally need to try different brands of medication before finding one that works specifically for them. Research shows that 1 in 4 of those who switch medications get better results regardless of whether the second medication is an SSRI or some other type of antidepressant.
In a meta-analytic study from 2005, it was found that SSRIs and TCAs are equally effective in treating dysthymia. They also found that MAOIs have a slight advantage over the use of other medication in treating this disorder. However, the author of this study cautions that MAOIs should not necessarily be the first line of defence in the treatment of dysthymia, as they are often less tolerable than their counterparts, such as SSRIs.
Tentative evidence supports the use of amisulpride to treat dysthymia but with increased side effects.
Combination Treatment
When pharmacotherapy alone is compared with combined treatment with pharmacotherapy plus psychotherapy, there is a strong trend in favour of combined treatment. Working with a psychotherapist to address the causes and effects of the disorder, in addition to taking antidepressants to help eliminate the symptoms, can be extremely beneficial. This combination is often the preferred method of treatment for those who have dysthymia. Looking at various studies involving treatment for dysthymia, 75% of people responded positively to a combination of cognitive behavioural therapy and pharmacotherapy, whereas only 48% of people responded positively to just CBT or medication alone.
A 2019 Cochrane review of 10 studies involving 840 participants could not conclude with certainty that continued pharmacotherapy with antidepressants (those used in the studies) was effective in preventing relapse or recurrence of persistent depressive disorder. The body of evidence was too small for any greater certainty although the study acknowledges that continued psychotherapy may be beneficial when compared to no treatment.
Resistance
Because of dysthymia’s chronic nature, treatment resistance is somewhat common. In such a case, augmentation is often recommended. Such treatment augmentations can include lithium pharmacology, thyroid hormone augmentation, amisulpride, buspirone, bupropion, stimulants, and mirtazapine. Additionally, if the person also suffers from seasonal affective disorder, light therapy can be useful in helping augment therapeutic effects.
Depression is a symptom of some physical diseases; a side effect of some drugs and medical treatments; and a symptom of some mood disorders such as major depressive disorder or dysthymia. Physical causes are ruled out with a clinical assessment of depression that measures vitamins, minerals, electrolytes, and hormones. Management of depression may involve a number of different therapies: medications, behaviour therapy, psychotherapy, and medical devices.
Though psychiatric medication is the most frequently prescribed therapy for major depression, psychotherapy may be effective, either alone or in combination with medication. Combining psychotherapy and antidepressants may provide a “slight advantage”, but antidepressants alone or psychotherapy alone are not significantly different from other treatments, or “active intervention controls”. Given an accurate diagnosis of major depressive disorder, in general the type of treatment (psychotherapy and/or antidepressants, alternate or other treatments, or active intervention) is “less important than getting depressed patients involved in an active therapeutic program.”
Psychotherapy is the treatment of choice in those under the age of 18, with medication offered only in conjunction with the former and generally not as a first line agent. The possibility of depression, substance misuse or other mental health problems in the parents should be considered and, if present and if it may help the child, the parent should be treated in parallel with the child.
Psychotherapy and Behaviour Therapy
There are a number of different psychotherapies for depression which are provided to individuals or groups by psychotherapists, psychiatrists, psychologists, clinical social workers, counsellors or psychiatric nurses. With more chronic forms of depression, the most effective treatment is often considered to be a combination of medication and psychotherapy. Psychotherapy is the treatment of choice in people under 18. A meta-analysis examined the effectiveness of psychotherapy for depression across ages from younger than 13 years to older than 75 years. It summarizes results from 366 trials included 36,702 patients. It found that the best results were for young adults, with an average effect size of g=.98 (95% CI, 0.79-1.16). The effects were smallest for young children (<13 years), g = .35 (95% CI, 0.15-0.55), and second largest in the oldest group, g = .97 (95% CI, 0.42-1.52). The study was not able to compare the different types of therapy to each other. Most of the studies with children used therapies originally developed with adults, which may have reduced the effectiveness. The greater benefits with young adults might be due to a large number of studies including college students, who might have an easier time learning therapy skills and techniques. Most of the studies in children were done in the USA, whereas in older age groups, more balanced numbers of studies came from Europe and other parts of the world as well.
As the most studied form of psychotherapy for depression, cognitive behavioural therapy (CBT) is thought to work by teaching clients to learn a set of cognitive and behavioural skills, which they can employ on their own. Earlier research suggested that cognitive behavioural therapy was not as effective as antidepressant medication in the treatment of depression; however, more recent research suggests that it can perform as well as antidepressants in treating patients with moderate to severe depression. Beck’s treatment manual, Cognitive therapy of depression, has undergone the most research and accumulated the most evidence for its use. However, a number of other CBT manuals also have evidence to support their effectiveness with depression.
The effect of psychotherapy on patient and clinician rated improvement as well as on revision rates have declined steadily from the 1970s.
A systematic review of data comparing low-intensity CBT (such as guided self-help by means of written materials and limited professional support, and website-based interventions) with usual care found that patients who initially had more severe depression benefited from low-intensity interventions at least as much as less-depressed patients.
For the treatment of adolescent depression, one published study found that CBT without medication performed no better than a placebo, and significantly worse than the antidepressant fluoxetine. However, the same article reported that CBT and fluoxetine outperformed treatment with only fluoxetine. Combining fluoxetine with CBT appeared to bring no additional benefit in two different studies or, at the most, only marginal benefit, in a fourth study.
Behaviour therapy for depression is sometimes referred to as behavioural activation. Studies exist showing behavioural activation to be superior to CBT. In addition, behavioural activation appears to take less time and lead to longer lasting change. Two well-researched treatment manuals include Social skills training for depression and Behavioural activation treatment for depression.
Emotionally focused therapy, founded by Sue Johnson and Les Greenberg in 1985, treats depression by identifying and processing underlying emotions. The treatment manual, Facilitating emotional change, outlines treatment techniques.
Acceptance and commitment therapy (ACT), a mindfulness form of CBT, which has its roots in behaviour analysis, also demonstrates that it is effective in treating depression, and can be more helpful than traditional CBT, especially where depression is accompanied by anxiety and where it is resistant to traditional CBT.
A review of four studies on the effectiveness of mindfulness-based cognitive therapy (MBCT), a recently developed class-based program designed to prevent relapse, suggests that MBCT may have an additive effect when provided with the usual care in patients who have had three or more depressive episodes, although the usual care did not include antidepressant treatment or any psychotherapy, and the improvement observed may have reflected non-specific or placebo effects. Of note, although Mindfulness-based cognitive therapy for depression prevented relapse of future depressive episodes, there is no research on whether it can cause the remission of a current depressive episode.
Interpersonal psychotherapy (IPT) focuses on the social and interpersonal triggers that may cause depression. There is evidence that it is an effective treatment for depression. Here, the therapy takes a fairly structured course (often 12 sessions, as in the original research versions) as in the case with CBT; however, the focus is on relationships with others. Unlike family therapy, IPT is an individual format, so it is possible to work on interpersonal themes even if other family members do not come to the session. Therapy can be used to help a person develop or improve interpersonal skills in order to allow him or her to communicate more effectively and reduce stress. In a meta-analysis of 16 studies and 4,356 patients, the average improvement in depressive symptoms was an effect size of d = 0.63 (95% CI, 0.36 to 0.90). IPT combined with pharmacotherapy was more effective in preventing relapse than pharmacotherapy alone, number needed to treat = 7.63.
Psychoanalysis, a school of thought founded by Sigmund Freud that emphasizes the resolution of unconscious mental conflicts, is used by its practitioners to treat clients presenting with major depression. A more widely practiced technique, called psychodynamic psychotherapy, is loosely based on psychoanalysis and has an additional social and interpersonal focus. In a meta-analysis of three controlled trials, psychodynamic psychotherapy was found to be as effective as medication for mild to moderate depression.
Shared Care
Shared decision making is an approach whereby patients and clinicians freely share important evidence when tasked with decision making and where patients are guided to consider the best available options to make an informed decision. The principles are well documented, but there is a gap in that it’s hard to apply them in routine clinical practice. The steps have been simplified into five steps. The first step is seeking patient participation in that the health practitioner is tasked with communicating existing choices and therefore inviting them to the decision making process. The next step involves assisting the patient to explore and compare the treatment options by a critical analysis of the risks and benefits. The third step involves the assessment of the patient’s values and what they prefer taking to account what is of paramount urgency to the patient. Step 4 involves decision making where the patient and the practitioner make a conclusive decision on the best option and arrange for subsequent follow up meetings. Finally, the fifth step involves the analysis of the patient’s decision’. Five steps for you and your patients to work together to make the best possible health care decisions. The step involves monitoring of the degree of implementation, overcoming of barriers of decision implantation consequently the decisions need to be revisited and optimised thus ensuring the decision has a positive impact on health outcomes its success relies on the ability of the health practitioner to create a good interpersonal relationship with the patient.
Depression still remains a major problem in the US whereby statistics have it that 16 million people were affected in the year 2017. The depression is multifactorial and has been on the increase due to societal pressure, genetic association and increase in use of drugs. incorporation of nursing in management of depression may seem important in that nursing holds a pivotal role in health care delivery where they are the health practitioners that have been trained to be versatile from clinical to psychological care. Their incorporation in shared decision making in treating depression may be important as nurses are known to have the best interpersonal relationship with the patients thus a better collaborative model can be achieved due to this fact. With this in mind, the nurses may serve to administer drugs in management, prepare and maintain the patient’s records, interaction with other care staff to achieve optimum care, and organising therapy sessions. In a study another study concerning shared decision-making interventions for people with mental health conditions there were no overt benefits that were discovered and the called for further research in this area. Another study found that it is important to begin the dissemination and implementation of SDM as they proved that it has benefits in healthcare especially in mental health care and has received social and government support and however transitioning to SDM has proven to be an uphill task. It has been suggested that SDM is of importance in demonstrating patient preferences in decision making when there is no clear approach to treatment. In addition, numerous tools can be used to make the decision making the process easier these include the Controlled Preferences Scale that informs clinicians on how to actively involve patients
Commentators suggest that providers need to embrace shared decision making by making sure that patients participate actively in their management thus enabling the success of the model.
Medication
To find the most effective pharmaceutical drug treatment, the dosages of medications must often be adjusted, different combinations of antidepressants tried, or antidepressants changed. Norepinephrine reuptake inhibitor (NRIs) can be used as antidepressants. Selective serotonin reuptake inhibitors (SSRIs), such as sertraline (Zoloft, Lustral), escitalopram (Lexapro, Cipralex), fluoxetine (Prozac), paroxetine (Seroxat), and citalopram, are the primary medications considered, due to their relatively mild side effects and broad effect on the symptoms of depression and anxiety, as well as reduced risk in overdose, compared to their older tricyclic alternatives. Those who do not respond to the first SSRI tried can be switched to another. If sexual dysfunction is present prior to the onset of depression, SSRIs should be avoided. Another popular option is to switch to the atypical antidepressant bupropion (Wellbutrin) or to add bupropion to the existing therapy; this strategy is possibly more effective. It is not uncommon for SSRIs to cause or worsen insomnia; the sedating noradrenergic and specific serotonergic antidepressant (NaSSA) antidepressant mirtazapine (Zispin, Remeron) can be used in such cases. CBT for Insomnia can also help to alleviate the insomnia without additional medication. Venlafaxine (Effexor) from the SNRI class may be moderately more effective than SSRIs; however, it is not recommended as a first-line treatment because of the higher rate of side effects, and its use is specifically discouraged in children and adolescents. Fluoxetine is the only antidepressant recommended for people under the age of 18, though, if a child or adolescent patient is intolerant to fluoxetine, another SSRI may be considered. Evidence of effectiveness of SSRIs in those with depression complicated by dementia is lacking.
Tricyclic antidepressants (TCAs) have more side effects than SSRIs (but less sexual dysfunctions) and are usually reserved for the treatment of inpatients, for whom the tricyclic antidepressant amitriptyline, in particular, appears to be more effective. A different class of antidepressants, the monoamine oxidase inhibitors, have historically been plagued by questionable efficacy (although early studies used dosages now considered too low) and life-threatening adverse effects. They are still used only rarely, although newer agents of this class (RIMA), with a better side effect profile, have been developed.
In older patients TCAs and SSRIs are of the same efficacy. However, there are differences between TCA related antidepressants and classical TCAs in terms of side effect profiles and withdrawal when compared to SSRIs.
There is evidence a prominent side-effect of antidepressants, emotional blunting, is confused with a symptom of depression itself. The cited study, according to Professor Linda Gask was: ‘funded by a pharmaceutical company (Servier) and two of its authors are employees of that company’, which may bias the results. The study authors’ note: “emotional blunting is reported by nearly half of depressed patients on antidepressants and that it appears to be common to all monoaminergic antidepressants not only SSRIs”. Additionally, they note: “The OQuESA scores are highly correlated with the HAD depression score; emotional blunting cannot be described simply as a side-effect of antidepressant, but also as a symptom of depression…More emotional blunting is associated with a poorer quality of remission…”
Acetyl-l-Carnitine
Acetylcarnitine levels were lower in depressed patients than controls and in rats it causes rapid antidepressant effects through epigenetic mechanisms. A systematic review and meta-analysis of 12 randomised controlled trials found “supplementation significantly decreases depressive symptoms compared with placebo/no intervention, while offering a comparable effect with that of established antidepressant agents with fewer adverse effects.”
Zinc
A 2012 cross-sectional study found an association between zinc deficiency and depressive symptoms among women, but not men, and a 2013 meta-analysis of 17 observational studies found that blood zinc concentrations were lower in depressed subjects than in control subjects. A 2012 meta-analysis found that zinc supplementation as an adjunct to antidepressant drug treatment significantly lowered depressive symptom scores of depressed patients. The potential mechanisms underlying the association between low serum zinc and depression remain unclear, but may involve the regulation of neurotransmitter, endocrine and neurogenesis pathways. Zinc supplementation has been reported to improve symptoms of ADHD and depression. A 2013 review found that zinc supplementation may be an effective treatment in major depression.
Magnesium
Many studies have found an association between magnesium intake and depression. Magnesium was lower in serum of depressed patients than controls. One trial found magnesium chloride to be effective for depression in seniors with type 2 diabetes while another trial found magnesium citrate decreased depression in patients with fibromyalgia. One negative trial used magnesium oxide, which is poorly absorbed. A randomised, open-label study found that consumption of magnesium chloride for 6 weeks resulted in a clinically significant net improvement in depression, and that effects were observed within 2 weeks.
Augmentation
Physicians often add a medication with a different mode of action to bolster the effect of an antidepressant in cases of treatment resistance; a 2002 large community study of 244,859 depressed Veterans Administration patients found that 22% had received a second agent, most commonly a second antidepressant. Lithium has been used to augment antidepressant therapy in those who have failed to respond to antidepressants alone. Furthermore, lithium dramatically decreases the suicide risk in recurrent depression. Addition of atypical antipsychotics when the patient has not responded to an antidepressant is also known to increase the effectiveness of antidepressant drugs, albeit at the cost of more frequent and potentially serious side effects. There is some evidence for the addition of a thyroid hormone, triiodothyronine, in patients with normal thyroid function. Stephen M. Stahl, renowned academician in psychopharmacology, has stated resorting to a dynamic psychostimulant, in particular, d-amphetamine is the “classical augmentation strategy for treatment-refractory depression”. However, the use of stimulants in cases of treatment-resistant depression is relatively controversial.
Efficacy of Medication and Psychotherapy
Antidepressants are statistically superior to placebo but their overall effect is low-to-moderate. In that respect they often did not exceed the National Institute for Health and Clinical Excellence (NICE) criteria for a “clinically significant” effect. In particular, the effect size was very small for moderate depression but increased with severity, reaching “clinical significance” for very severe depression. These results were consistent with the earlier clinical studies in which only patients with severe depression benefited from either psychotherapy or treatment with an antidepressant, imipramine, more than from the placebo treatment. Despite obtaining similar results, the authors argued about their interpretation. One author concluded that there “seems little evidence to support the prescription of antidepressant medication to any but the most severely depressed patients, unless alternative treatments have failed to provide benefit.” The other author agreed that “antidepressant ‘glass’ is far from full” but disagreed “that it is completely empty”. He pointed out that the first-line alternative to medication is psychotherapy, which does not have superior efficacy.
Antidepressants in general are as effective as psychotherapy for major depression, and this conclusion holds true for both severe and mild forms of MDD. In contrast, medication gives better results for dysthymia. The subgroup of SSRIs may be slightly more efficacious than psychotherapy. On the other hand, significantly more patients drop off from the antidepressant treatment than from psychotherapy, likely because of the side effects of antidepressants. Successful psychotherapy appears to prevent the recurrence of depression even after it has been terminated or replaced by occasional “booster” sessions. The same degree of prevention can be achieved by continuing antidepressant treatment.
Two studies suggest that the combination of psychotherapy and medication is the most effective way to treat depression in adolescents. Both TADS (Treatment of Adolescents with Depression Study) and TORDIA (Treatment of Resistant Depression in Adolescents) showed very similar results. TADS resulted in 71% of their teen subjects having “much” or “very much” improvement in mood over the 61% with medication alone and 43% with CBT alone. Similarly, TORDIA showed a 55% improvement with CBT and drugs versus a 41% with drug therapy alone. However, a more recent meta-analysis of 34 trials of 14 drugs used with children and adolescents found that only fluoxetine produced significant benefit compared to placebo, with a medium sized effect (standardize mean difference = .5).
Treatment Resistance
The risk factors for treatment resistant depression are: the duration of the episode of depression, severity of the episode, if bipolar, lack of improvement in symptoms within the first couple of treatment weeks, anxious or avoidant and borderline comorbidity and old age. Treatment resistant depression is best handled with a combination of conventional antidepressant together with atypical antipsychotics. Another approach is to try different antidepressants. It is inconclusive which approach is superior. Treatment resistant depression can be misdiagnosed if subtherapeutic doses of antidepressants is the case, patient nonadherence, intolerable adverse effects or their thyroid disease or other conditions is misdiagnosed as depression.
Experimental Treatments
Chromium
Clinical and experimental studies have reported antidepressant activity of chromium particularly in atypical depression, characterised by increased appetite and carbohydrate craving.
Essential Fatty Acids
A 2015 Cochrane Collaboration review found insufficient evidence with which to determine if omega-3 fatty acid has any effect on depression. A 2016 review found that if trials with formulations containing mostly eicosapentaenoic acid (EPA) are separated from trials using formulations containing docosahexaenoic acid (DHA), it appeared that EPA may have an effect while DHA may not, but there was insufficient evidence to be sure.
Creatine
The amino acid creatine, commonly used as a supplement to improve the performance of bodybuilders, has been studied for its potential antidepressant properties. A double-blinded, placebo-controlled trial focusing on women with major depressive disorder found that daily creatine supplementation adjunctive to escitalopram was more effective than escitalopram alone. Studies on mice have found that the antidepressant effects of creatine can be blocked by drugs that act against dopamine receptors, suggesting that the drug acts on dopamine pathways.
Dopamine Receptor Agonist
Some research suggests dopamine receptor agonist may be effective in treating depression, however studies are few and results are preliminary.
Inositol
Inositol, an alcohol sugar found in fruits, beans grains and nuts may have antidepressant effects in high doses. Inositol may exert its effects by altering intracellular signalling.
Ketamine
Research on the antidepressant effects of ketamine infusions at subanaesthetic doses has consistently shown rapid (4 to 72 hours) responses from single doses, with substantial improvement in mood in the majority of patients and remission in some. However, these effects are often short-lived, and attempts to prolong the antidepressant effect with repeated doses and extended (“maintenance”) treatment have resulted in only modest success.
N-Acetylcysteine
A systematic review and meta-analysis of 5 studies found that N-Acetylcysteine reduces depressive symptoms more than placebo and has good tolerability. N-Acetylecysteine may exert benefits as a precursor to the antioxidant glutathione, thus modulating glutamatergic, neurotropic, and inflammatory pathways.
St John’s Wort
A 2008 Cochrane Collaboration meta-analysis concluded that:
“The available evidence suggests that the hypericum extracts tested in the included trials a) are superior to placebo in patients with major depression; b) are similarly effective as standard antidepressants; c) and have fewer side effects than standard antidepressants. The association of country of origin and precision with effects sizes complicates the interpretation.”
The United States National Centre for Complementary and Integrative Health advice is that “St. John’s wort may help some types of depression, similar to treatment with standard prescription antidepressants, but the evidence is not definitive.” and warns that “Combining St. John’s wort with certain antidepressants can lead to a potentially life-threatening increase of serotonin, a brain chemical targeted by antidepressants. St. John’s wort can also limit the effectiveness of many prescription medicines.”
Rhodiola Rosea
A 2011 review reported Rhodiola rosea “is an adaptogen plant that can be especially helpful in treating asthenic or lethargic depression, and may be combined with conventional antidepressants to alleviate some of their common side effects.” A 6 week double-blind, placebo-controlled, randomised study with 89 patients with mild to moderate depression found that R. rosea statistically significantly reduced depression symptoms, and no side effects were reported.
Saffron
A 2013 meta-analysis found that saffron supplementation significantly reduced depression symptoms compared to placebo, and both saffron supplementation and the antidepressant groups were similarly effective in reducing depression symptoms. A 2015 meta-analysis supported the “efficacy of saffron as compared to placebo in improving the following conditions: depressive symptoms (compared to anti-depressants and placebo), premenstrual symptoms, and sexual dysfunction. In addition, saffron use was also effective in reducing excessive snacking behavior.” The antidepressant effect of saffron stigma extracts may be mediated via its components safranal and crocin: “crocin may act via the uptake inhibition of dopamine and norepinephrine, and safranal via serotonin.” Therapeutic doses of saffron exhibits no significant toxicity in both clinical and experimental investigations.
SAMe
S-Adenosyl methionine (SAMe) is available as a prescription antidepressant in Europe and an over-the-counter dietary supplement in the US. Evidence from 16 clinical trials with a small number of subjects, reviewed in 1994 and 1996 suggested it to be more effective than placebo and as effective as standard antidepressant medication for the treatment of major depression.
Tryptophan and 5-HTP
The amino acid tryptophan is converted into 5-hydroxytryptophan (5-HTP) which is subsequently converted into the neurotransmitter serotonin. Since serotonin deficiency has been recognized as a possible cause of depression, it has been suggested that consumption of tryptophan or 5-HTP may therefore improve depression symptoms by increasing the level of serotonin in the brain. 5-HTP and tryptophan are sold over the counter in North America, but requires a prescription in Europe. The use of 5-HTP instead of tryptophan bypasses the conversion of tryptophan into 5-HTP by the enzyme tryptophan hydroxylase, which is the rate-limiting step in the synthesis of serotonin, and 5-HTP easily crosses the blood–brain barrier unlike tryptophan, which requires a transporter.
Small studies have been performed using 5-HTP and tryptophan as adjunctive therapy in addition to standard treatment for depression. While some studies had positive results, they were criticised for having methodological flaws, and a more recent study did not find sustained benefit from their use. The safety of these medications has not been well studied. Due to the lack of high quality studies, preliminary nature of studies showing effectiveness, the lack of adequate study on their safety, and reports of Eosinophilia-myalgia syndrome from contaminated tryptophan in 1989 and 1990, the use of tryptophan and 5-HTP is not highly recommended or thought to be clinically useful.
Medical Devices
A variety of medical devices are in use or under consideration for treatment of depression including devices that offer electroconvulsive therapy, vagus nerve stimulation, repetitive transcranial magnetic stimulation, and cranial electrotherapy stimulation. The use of such devices in the United States requires approval by the US Food and Drug Administration (FDA) after field trials. In 2010 an FDA advisory panel considered the question of how such field trials should be managed. Factors considered were whether drugs had been effective, how many different drugs had been tried, and what tolerance for suicides should be in field trials.
Electroconvulsive Therapy
Electroconvulsive therapy (ECT) is a standard psychiatric treatment in which seizures are electrically induced in patients to provide relief from psychiatric illnesses. ECT is used with informed consent as a last line of intervention for major depressive disorder. Among the elderly, who often experience depression, the efficacy of ECT is difficult to determine due to the lack of trials comparing ECT to other treatments.
A round of ECT is effective for about 50% of people with treatment-resistant major depressive disorder, whether it is unipolar or bipolar. Follow-up treatment is still poorly studied, but about half of people who respond, relapse with twelve months.
Aside from effects in the brain, the general physical risks of ECT are similar to those of brief general anaesthesia. Immediately following treatment, the most common adverse effects are confusion and memory loss. ECT is considered one of the least harmful treatment options available for severely depressed pregnant women.
A usual course of ECT involves multiple administrations, typically given two or three times per week until the patient is no longer suffering symptoms ECT is administered under anaesthetic with a muscle relaxant. Electroconvulsive therapy can differ in its application in three ways: electrode placement, frequency of treatments, and the electrical waveform of the stimulus. These three forms of application have significant differences in both adverse side effects and symptom remission. After treatment, drug therapy is usually continued, and some patients receive maintenance ECT.
ECT appears to work in the short term via an anticonvulsant effect mostly in the frontal lobes, and longer term via neurotrophic effects primarily in the medial temporal lobe.
Deep Brain Stimulation
The support for the use of deep brain stimulation in treatment-resistant depression comes from a handful of case studies, and this treatment is still in a very early investigational stage. In this technique electrodes are implanted in a specific region of the brain, which is then continuously stimulated. A March 2010 systematic review found that “about half the patients did show dramatic improvement” and that adverse events were “generally trivial” given the younger psychiatric patient population than with movements disorders. Deep brain stimulation is available on an experimental basis only in the United States; no systems are approved by the FDA for this use. It is available in Australia.
Repetitive Transcranial Magnetic Stimulation
Transcranial magnetic stimulation (TMS) or deep transcranial magnetic stimulation is a non-invasive method used to stimulate small regions of the brain. During a TMS procedure, a magnetic field generator, or “coil” is placed near the head of the person receiving the treatment. The coil produces small electric currents in the region of the brain just under the coil via electromagnetic induction. The coil is connected to a pulse generator, or stimulator, that delivers electric current to the coil.
TMS was approved by the FDA for treatment-resistant major depressive disorder in 2008 and as of 2014 clinical evidence supports this use. The American Psychiatric Association, the Canadian Network for Mood and Anxiety Disorders, and the Royal Australia and New Zealand College of Psychiatrists have endorsed rTMS for trMDD.
Vagus Nerve Stimulation
Vagus nerve stimulation (VNS) uses an implanted electrode and generator to deliver electrical pulses to the vagus nerve, one of the primary nerves emanating from the brain. It is an approved therapy for treatment-resistant depression in the EU and US and is sometimes used as an adjunct to existing antidepressant treatment. The support for this method comes mainly from open-label trials, which indicate that several months may be required to see a benefit. The only large double-blind trial conducted lasted only 10 weeks and yielded inconclusive results; VNS failed to show superiority over a sham treatment on the primary efficacy outcome, but the results were more favourable for one of the secondary outcomes. The authors concluded “This study did not yield definitive evidence of short-term efficacy for adjunctive VNS in treatment-resistant depression.”
Cranial Electrotherapy Stimulation
A 2014 Cochrane review found insufficient evidence to determine whether or not Cranial electrotherapy stimulation with alternating current is safe and effective for treating depression.
Transcranial Direct Current Stimulation
A 2016 meta-analysis of transcranial direct current stimulation (tDCS) reported some efficacy of tDCS in the treatment of acute depressive disorder with moderate effect size, and low efficacy in treatment-resistant depression, and that use of 2 mA current strength over 20 minutes per day over a short time span can be considered safe.
Other Treatments
Bright Light Therapy
A meta-analysis of bright light therapy commissioned by the American Psychiatric Association found a significant reduction in depression symptom severity associated with bright light treatment. Benefit was found for both seasonal affective disorder and for non-seasonal depression, with effect sizes similar to those for conventional antidepressants. For non-seasonal depression, adding light therapy to the standard antidepressant treatment was not effective. A meta-analysis of light therapy for non-seasonal depression conducted by Cochrane Collaboration, studied a different set of trials, where light was used mostly in combination with antidepressants or wake therapy. A moderate statistically significant effect of light therapy was found, with response significantly better than control treatment in high-quality studies, in studies that applied morning light treatment, and with patients who respond to total or partial sleep deprivation. Both analyses noted poor quality of most studies and their small size, and urged caution in the interpretation of their results. The short 1-2 weeks duration of most trials makes it unclear whether the effect of light therapy could be sustained in the longer term.
Exercise
The 2013 Cochrane Collaboration review on physical exercise for depression noted that, based upon limited evidence, it is moderately more effective than a control intervention and comparable to psychological or antidepressant drug therapies. Smaller effects were seen in more methodologically rigorous studies. Three subsequent 2014 systematic reviews that included the Cochrane review in their analysis concluded with similar findings: one indicated that physical exercise is effective as an adjunct treatment with antidepressant medication; the other two indicated that physical exercise has marked antidepressant effects and recommended the inclusion of physical activity as an adjunct treatment for mild-moderate depression and mental illness in general. These studies also found smaller effect sizes in more methodologically rigorous studies. All four systematic reviews called for more research in order to determine the efficacy or optimal exercise intensity, duration, and modality. The evidence for brain-derived neurotrophic factor (BDNF) in mediating some of the neurobiological effects of physical exercise was noted in one review which hypothesized that increased BDNF signalling is responsible for the antidepressant effect.
Meditation
Mindfulness meditation programs may help improve symptoms of depression, but they are no better than active treatments such as medication, exercise, and other behavioural therapies.
Music Therapy
A 2009 review found that 3 to 10 sessions of music therapy resulted in a noticeable improvement in depressive symptoms, with still greater improvement after 16 to 51 sessions.
Sleep
Depression is sometimes associated with insomnia – (difficulty in falling asleep, early waking, or waking in the middle of the night). The combination of these two results, depression and insomnia, will only worsen the situation. Hence, good sleep hygiene is important to help break this vicious circle. It would include measures such as regular sleep routines, avoidance of stimulants such as caffeine and management of sleeping disorders such as sleep apnoea.
Smoking Cessation
Quitting smoking cigarettes is associated with reduced depression and anxiety, with the effect “equal or larger than” those of antidepressant treatments.
Total/Partial Sleep Deprivation
Sleep deprivation (skipping a night’s sleep) has been found to improve symptoms of depression in 40-60% of patients. Partial sleep deprivation in the second half of the night may be as effective as an all night sleep deprivation session. Improvement may last for weeks, though the majority (50-80%) relapse after recovery sleep. Shifting or reduction of sleep time, light therapy, antidepressant drugs, and lithium have been found to potentially stabilise sleep deprivation treatment effects.
Shared Care
Shared care, when primary and specialty physicians have joint management of an individual’s health care, has been shown to alleviate depression outcomes.
The Centre for Epidemiologic Studies Depression Scale (CES-D) is a brief self-report questionnaire developed in 1977 by Laurie Radloff to measure depressive symptoms severity in the general population.
The CES-D consists of 20 questions that asks about various symptoms of depression as they have occurred in the past week, and the majority of the items focus on the affective component of depression. Although initially designed for use in general population surveys, CES-D now serves as a screening instrument in primary care clinics and in research.
A revision, the CESD-R was produced in 2004.
Centre for Epidemiologic Studies Depression Scale for Children
The Centre for Epidemiologic Studies Depression Scale for Children (CES-DC) is a modified version of the Centre for Epidemiologic Studies Depression Scale. This measure assesses both depressive symptoms as well as symptom improvement in a wide range of children and adolescents, ages 6-17. The CES-DC was first developed to measure the incidence and prevalence of depression among children and adolescents in large-scale epidemiological research. Several research studies have found the CES-DC to be a reliable and valid measure of depressive symptoms in children.
Question Breakdown and Scoring
The CES-DC is an inventory of 20 self-report items regarding depressive symptoms, taking about 5 minutes to complete. Each item asks how often a symptom has occurred within the last week. Response choices are assigned point values, which are summed together to determine a total measure score. Response choices for each item and their corresponding point values are as follows:
0 points: “Not at all”.
1 point: “A little”.
2 points: “Some”.
3 points: “A lot”.
Items 4, 8, 12 and 16 are phrased to reflect positive affect and behaviour, and therefore are scored in opposite order as follows:
0 points: “A lot”.
1 point: “Some”.
2 points: “A little”.
3 points: “Not at all”.
Interpretation
Scores on the CES-DC range from 0 to 60, in which higher scores suggest a greater presence of depressive symptoms. A score of 15 or higher is interpreted to indicate a risk for depression. However, screening for depression is a complex process and scoring a 15 or higher on the CES-DC should be followed by further evaluation.
Limitations
A study evaluating the CES-DC found that the scores do not necessarily match up to a DSM diagnosis, and while it is a good psychometric tool for adolescents, reliability and validity is poor when applied to children.
The Mood and Feelings Questionnaire (MFQ) is a survey that measures depressive symptoms in children and young adults.
Background
It was developed by Adrian Angold and Elizabeth J. Costello in 1987, and validity data were gathered as part of the Great Smokey Mountain epidemiological study in Western North Carolina.
The questionnaire consists of a variety of statements describing feelings or behaviours that may manifest as depressive symptoms in children between the ages of 6 and 17. The subject is asked to indicate how much each statement applies to their recent experiences. The Mood and Feelings Questionnaire has six versions, short (13 item) and long (33 item) forms of each of the following:
A youth self-report;
A version that a parent would complete; and
A self-report version for adults.
Several peer-reviewed studies have found the Mood and Feelings Questionnaire to be a reliable and valid measure of depression in children. Compared to many other depression scales for youth, it has more extensive coverage of symptoms and more age-appropriate wording and content.
Scoring and Interpretation
The MFQ has several tests, one short and one long, with the short questionnaire including 13 questions and the long questionnaire consisting of 33 questions. Scoring of the MFQ works by summing the point values allocated to each question. The responses and their allocated point values are as follows:
“not true” = 0 points.
“somewhat true” = 1 point.
“true” = 2 points.
Scores on the short MFQ range from 0 to 26, whereas scores on the long version range from 0 to 66. Higher score are indicative of increased depressive symptom severity. Scores larger than 12 on the short version or larger than 27 on the long version are suggestive of likely depression and warrant further clinical assessment.
Validity
The Mood and Feelings Questionnaire, along with the Short Mood and Feelings Questionnaire, shows reasonable psychometric properties for identifying children in early adolescence with a depressive disorder. Secondly, the MFQ does not significantly differentiate between children with depression versus children with anxiety disorders. Finally, the MFQ has been translated into Arabic, Spanish and Norwegian, but testing of these versions is more limited.
Limitations
Questionnaires like the Mood and Feelings Questionnaire should not act as a substitute for thorough clinical evaluations for both the child and parent.
Recurrent brief depression (RBD) defines a mental disorder characterised by intermittent depressive episodes, not related to menstrual cycles in women, occurring between approximately 6-12 times per year, over at least one year or more fulfilling the diagnostic criteria for major depressive episodes (DSM-IV and ICD-10) except for duration which in RBD is less than 14 days, typically 5-7 days.
Despite the short duration of the depressive episodes, such episodes are severe, and suicidal ideation and impaired function is rather common. The majority of patients with RBD also report symptoms of anxiety and increased irritability. Hypersomnia is also rather frequent. About 1/2 of patients fulfilling diagnostic criteria for RBD may have additional short episodes of brief hypomania which is a severity marker of RBD. RBD may be the only mental disorder present, but RBD may also occur as part of a history of recurrent major depressive episodes or bipolar disorders. RBD is also seen among some patients with personality disorders.
Prevalence
The lifetime prevalence of RBD has been estimated at 2.6-10.0%, and the one-year prevalence at 5.0-8.2%. The WHO project on “Psychological problems in general health care”, which was based on primary care samples, reported a one-year prevalence of 3.7-9.9%. However none of these studies differentiate between RBD with and without a history of other mood disorders (e.g. major depression). DSM-IV field trial estimated the lifetime of RBD only to be about 2%.
Brief History
Disorders characterised by periods with depressive episodes lasting hours to days have been described since 1852 and have been labelled “periodic melancholia”, “intermittent depressive disorder” or “very brief depression”. The third version of the Diagnostic and Statistical Manual of Mental Disorders (1980), which relied heavily on findings from studies conducted in psychiatric in- and out-patient settings, required at least 14 days duration for a diagnosis of depression. No diagnostic category was allocated a depressive episode of shorter duration. Thus, intermittent depressive disorder, included in the Research Diagnostic Criteria (1975) was considered to identify minor versions of major depression (“minor depression”) and not included in the DSM-III.
However, based on data from epidemiological studies, the Swiss psychiatrist and researcher, Jules Angst, coined the concept “recurrent brief depression” (RBD) and provided diagnostic criteria for this type of mood disorder in 1985. Several other European studies independently confirmed the occurrence of RBD in the general population and clinical samples. RBD was thus included in the 10th classification of mental and behavioural disorders (ICD-10 F38.1) published by the World Health Organisation in 1992 (WHO, 1992; WHO, 1993). Less frequent episodes of brief depressions were labelled infrequent brief depression and not included in ICD-10. The American classification system of mental disorders, DSM-IV (1994), provided provisional diagnostic criteria for RBD, but decided to await further studies before including RBD in the classification system. The fate of RBD in DSM-5, expected to occur in 2013, is not known.
Causes
The cause (aetiology) of RBD is unknown, but recent findings may suggest a link between RBD and bipolar disorders, pointing to the importance of genetic factors. A small subgroup of patients with RBD has temporal lobe epilepsy.
Diagnosis
From the International Statistical Classification of Mental and Behavioral Disorders:
F33 Recurrent depressive disorder
G1. There has been at least one previous episode, mild (F32.0), moderate (F32.1), or severe (F32.2 or F32.3), lasting a maximum of two weeks and separated from the current episode by at least two months free from any significant mood symptoms.
G2. At no time in the past has there been an episode meeting the criteria or hypomanic or manic episode (F30.-).
G3. Most commonly used exclusion criteria: the episode is not attributable to psychoactive substance use (F1) or any organic mental disorder, in the sense of F0. It is recommended to specify the predominant type of previous episodes (mild, moderate, severe, uncertain).
F33.0 Recurrent depressive disorder, current episode mild
A. The general criteria for recurrent depressive disorder (F33) are met.
B. The current episode meets the criteria for depressive episode, mild severity (F32.0).
A fifth character may be used to specify the presence of the somatic syndrome, as defined in F32, in the current episode:
F33.00 without somatic syndrome.
F33.01 with somatic syndrome.
F33.1 Recurrent depressive disorder, current episode moderate
A. The general criteria for recurrent depressive disorders (F33) are met.
B. The current episode meets the criteria for depressive episode, moderate severity (F32.1).
A fifth character may be used to specify the presence of the somatic syndrome, as defined in F32, in the current episode:
F33.10 without somatic syndrome
F33.11 with somatic syndrome.
F33.2 Recurrent depressive disorder, current episode severe without psychotic symptoms
A. The general criteria for recurrent depressive disorders (F33) are met.
B. The current episode meets the criteria for severe depressive episode without psychotic symptoms (F32.2).
F33.3 Recurrent depressive disorder, current episode severe with psychotic symptoms
A. The general criteria for recurrent depressive disorders (F33) are met.
B. The current episode meets the criteria for severe depressive episode with psychotic symptoms (F32.3). A fifth character may be used to specify whether the psychotic symptoms are congruent or incongruent with the mood:
F33.30 with mood congruent psychotic symptoms.
F33.31 with mood incongruent psychotic symptoms.
F33.4 Recurrent depressive disorder, currently in remission
A. The general criteria for recurrent depressive disorder (F33) have been met in the past.
B. The current state does not meet the criteria for a depressive episode (F32.-) of any severity, or for any other disorder in F3 (the patient may receive treatment to reduce the risk of further episodes).
F33.8 Other recurrent depressive disorders.
F33.9 Recurrent depressive disorder, unspecified.
Treatment
Both psychotherapy as well as different drugs (e.g. serotonin reuptake inhibitors – SSRIs or mood stabilisers, e.g. lithium, antiepileptics) have been suggested as treatments. However, no randomised controlled treatment trial of RBD has been conducted.
Disruption of long-term depression potentiates latent inhibition: Key role for central nucleus of the amygdala.
Background
Latent inhibition (LI) reflects an adaptive form of learning, which is impaired in certain forms of mental illness. Glutamate receptor activity is linked to LI, but the potential role of synaptic plasticity remains unspecified.
Methods
Accordingly, the present study examined the possible role of long-term depression (LTD) in LI induced by prior exposure of rats to an auditory stimulus used subsequently as a conditional stimulus (CS) to signal a pending footshock. The researchers employed two mechanistically distinct LTD inhibitors, the Tat-GluA23Y peptide that blocks endocytosis of the GluA2-containing glutamate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), or the selective glutamate n-methyl-d-aspartate receptor (NMDAR) 2B antagonist, Ro25-6981, administered prior to the acquisition of two-way conditioned avoidance with or without tone pre-exposure.
Results
Systemic LTD blockade with the Tat-GluA23Y peptide strengthened the LI effect by further impairing acquisition of conditioned avoidance in CS-pre-exposed rats compared to normal conditioning in non-pre-exposed controls. Systemic Ro25-6981 had no significant effects. Brain-region specific microinjections of the Tat-GluA23Y peptide into the nucleus accumbens, medial prefrontal cortex, central or basolateral amygdala demonstrated that disruption of AMPAR endocytosis in the central amygdala also potentiated the LI effect.
Conclusions
These data revealed a previously unknown role for central amygdala LTD in LI as a key mediator of cognitive flexibility required to respond to previously irrelevant stimuli that acquire significance through reinforcement. The findings may have relevance both for our mechanistic understanding of LI and its alteration in disease states such as schizophrenia, while further elucidating the role of LTD in learning and memory.
Reference
Ashby, D.M., Dias, C., Aleksandrova, L.R., Lapish, C.C., Wang, Y.T. & Phillips, A.G. (2021) Disruption of long-term depression potentiates latent inhibition: Key role for central nucleus of the amygdala. The International Journal of Neuropsychopharmacology. doi: 10.1093/ijnp/pyab011. Online ahead of print.



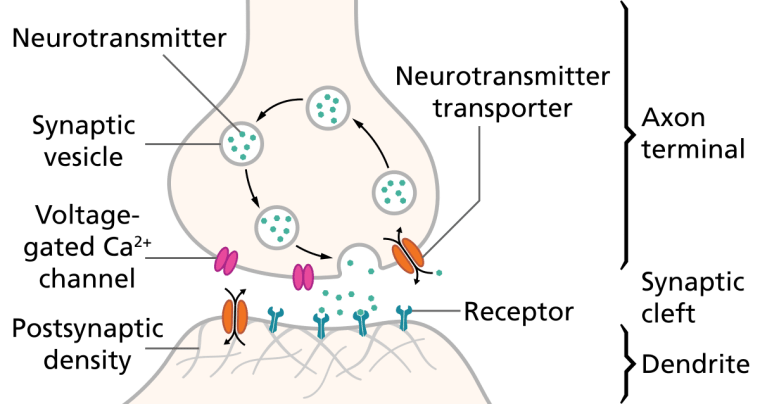
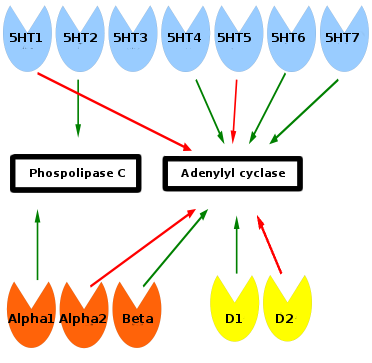
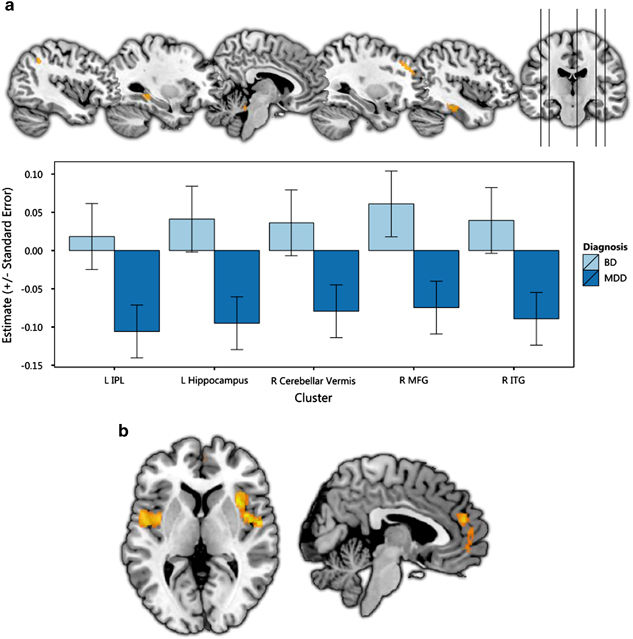


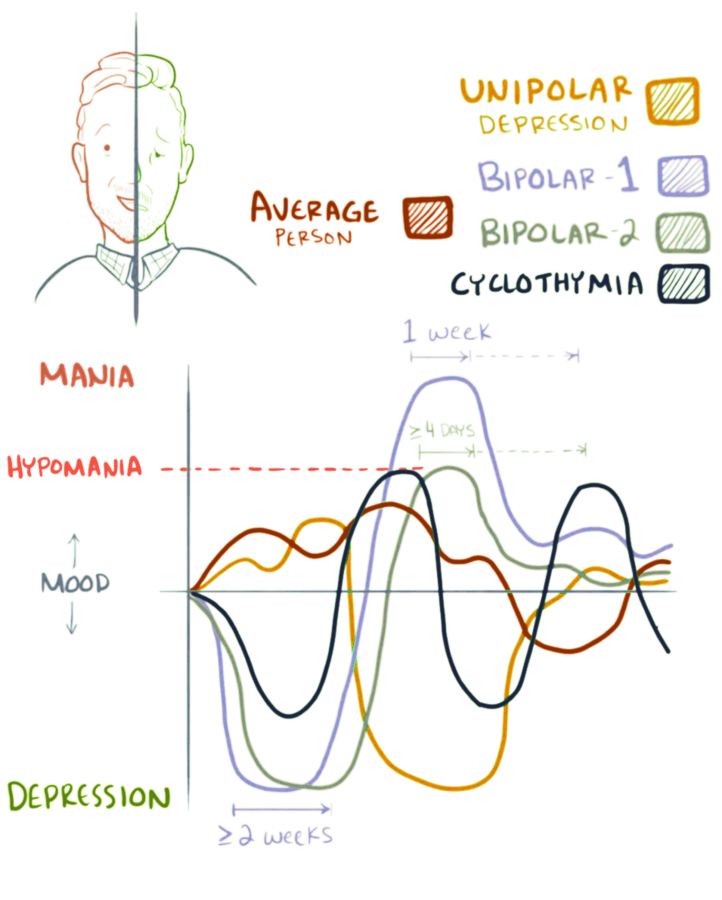


You must be logged in to post a comment.