Introduction
Temazepam, sold under the brand names Restoril among others, is a medication used to treat insomnia.
Such use should generally be for less than ten days. It is taken by mouth. Effects generally begin within an hour and last for up to eight hours.
Common side effects include sleepiness, anxiety, confusion, and dizziness. Serious side effects may include hallucinations, abuse, anaphylaxis, and suicide. Use is generally not recommended together with opioids. If the dose is rapidly decreased withdrawal may occur. Use during pregnancy or breastfeeding is not recommended. Temazepam is an intermediate acting benzodiazepine and hypnotic. It works by affecting GABA within the brain.
Temazepam was patented in 1962 and came into medical use in 1969. It is available as a generic medication. In 2017, it was the 142nd most commonly prescribed medication in the United States, with more than four million prescriptions.
Brief History
Temazepam was synthesized in 1964, but it came into use in 1981 when its ability to counter insomnia was realised. By the late 1980s, temazepam was one of the most popular and widely prescribed hypnotics on the market and it became one of the most widely prescribed drugs.
Medical Uses
In sleep laboratory studies, temazepam significantly decreased the number of nightly awakenings, but has the drawback of distorting the normal sleep pattern. It is officially indicated for severe insomnia and other severe or disabling sleep disorders. The prescribing guidelines in the UK limit the prescribing of hypnotics to two to four weeks due to concerns of tolerance and dependence.
The United States Air Force uses temazepam as one of the hypnotics approved as a “no-go pill” to help aviators and special-duty personnel sleep in support of mission readiness. “Ground tests” are necessary prior to required authorisation being issued to use the medication in an operational situation, and a 12-hour restriction is imposed on subsequent flight operation. The other hypnotics used as “no-go pills” are zaleplon and zolpidem, which have shorter mandatory recovery periods.
Contraindications
Use of temazepam should be avoided, when possible, in individuals with these conditions:
- Ataxia (gross lack of coordination of muscle movements).
- Severe hypoventilation.
- Acute narrow-angle glaucoma.
- Severe hepatic deficiencies (hepatitis and liver cirrhosis decrease elimination by a factor of two).
- Severe renal deficiencies (e.g. patients on dialysis).
- Sleep apnoea.
- Severe depression, particularly when accompanied by suicidal tendencies.
- Acute intoxication with alcohol, narcotics, or other psychoactive substances.
- Myasthenia gravis (autoimmune disorder causing muscle weakness).
- Hypersensitivity or allergy to any drug in the benzodiazepine class.
Special Caution Needed
Temazepam should not be used in pregnancy, as it may cause harm to the foetus. The safety and effectiveness of temazepam has not been established in children; therefore, temazepam should generally not be given to individuals under 18 years of age, and should not be used at all in children under six months old. Benzodiazepines also require special caution if used in the elderly, alcohol- or drug-dependent individuals, and individuals with comorbid psychiatric disorders.
Temazepam, similar to other benzodiazepines and nonbenzodiazepine hypnotic drugs, causes impairments in body balance and standing steadiness in individuals who wake up at night or the next morning. Falls and hip fractures are frequently reported. The combination with alcohol increases these impairments. Partial but incomplete tolerance develops to these impairments. The smallest possible effective dose should be used in elderly or very ill patients, as a risk of apnoea and/or cardiac arrest exists. This risk is increased when temazepam is given concomitantly with other drugs that depress the central nervous system (CNS).
Misuse and Dependence
Because benzodiazepines can be abused and lead to dependence, their use should be avoided in people in certain particularly high-risk groups. These groups include people with a history of alcohol or drug dependence, people significantly struggling with their mood or people with longstanding mental health difficulties. If temazepam must be prescribed to people in these groups, they should generally be monitored very closely for signs of misuse and development of dependence.
Adverse Effects
Refer to Benzodiazepine Withdrawal Syndrome.
In September 2020, the US Food and Drug Administration (FDA) required the boxed warning be updated for all benzodiazepine medicines to describe the risks of abuse, misuse, addiction, physical dependence, and withdrawal reactions consistently across all the medicines in the class.
Common
Side effects typical of hypnotic benzodiazepines are related to CNS depression, and include somnolence, sedation, dizziness, fatigue, ataxia, headache, lethargy, impairment of memory and learning, longer reaction time and impairment of motor functions (including coordination problems), slurred speech, decreased physical performance, numbed emotions, reduced alertness, muscle weakness, blurred vision (in higher doses), and inattention. Euphoria was rarely reported with its use. According to the FDA, temazepam had an incidence of euphoria of 1.5%, much more rarely reported than headaches and diarrhoea. Anterograde amnesia may also develop, as may respiratory depression in higher doses.
A 2009 meta-analysis found a 44% higher rate of mild infections, such as pharyngitis or sinusitis, in people taking Temazepam or other hypnotic drugs compared to those taking a placebo.
Less Common
Hyperhydrosis, hypotension, burning eyes, increased appetite, changes in libido, hallucinations, faintness, nystagmus, vomiting, pruritus, gastrointestinal disturbances, nightmares, palpitation and paradoxical reactions including restlessness, aggression, violence, overstimulation and agitation have been reported, but are rare (less than 0.5%).
Before taking temazepam, one should ensure that at least 8 hours are available to dedicate to sleep. Failing to do so can increase the side effects of the drug.
Like all benzodiazepines, the use of this drug in combination with alcohol potentiates the side effects, and can lead to toxicity and death.
Though rare, residual “hangover” effects after night-time administration of temazepam occasionally occur. These include sleepiness, impaired psychomotor and cognitive functions which may persist into the next day, impaired driving ability, and possible increased risks of falls and hip fractures, especially in the elderly.
Tolerance
Chronic or excessive use of temazepam may cause drug tolerance, which can develop rapidly, so this drug is not recommended for long-term use. In 1979, the US Institute of Medicine and the National Institute on Drug Abuse stated that most hypnotics lose their sleep-inducing properties after about three to 14 days. In use longer than one to two weeks, tolerance will rapidly develop towards the ability of temazepam to maintain sleep, resulting in a loss of effectiveness. Some studies have observed tolerance to temazepam after as little as one week’s use. Another study examined the short-term effects of the accumulation of temazepam over seven days in elderly inpatients, and found little tolerance developed during the accumulation of the drug. Other studies examined the use of temazepam over six days and saw no evidence of tolerance. A study in 11 young male subjects showed significant tolerance occurs to temazepam’s thermoregulatory effects and sleep inducing properties after one week of use of 30-mg temazepam. Body temperature is well correlated with the sleep-inducing or insomnia-promoting properties of drugs.
In one study, the drug sensitivity of people who had used temazepam for one to 20 years was no different from that of controls. An additional study, in which at least one of the authors is employed by multiple drug companies, examined the efficacy of temazepam treatment on chronic insomnia over three months, and saw no drug tolerance, with the authors even suggesting the drug might become more effective over time.
Establishing continued efficacy beyond a few weeks can be complicated by the difficulty in distinguishing between the return of the original insomnia complaint and withdrawal or rebound related insomnia. Sleep EEG studies on hypnotic benzodiazepines show tolerance tends to occur completely after one to four weeks with sleep EEG returning to pre-treatment levels. The paper concluded that due to concerns about long-term use involving toxicity, tolerance and dependence, as well as to controversy over long-term efficacy, wise prescribers should restrict benzodiazepines to a few weeks and avoid continuing prescriptions for months or years. A review of the literature found the nonpharmacological treatment options were a more effective treatment option for insomnia due to their sustained improvements in sleep quality.
Physical Dependence
Temazepam, like other benzodiazepine drugs, can cause physical dependence and addiction. Withdrawal from temazepam or other benzodiazepines after regular use often leads to benzodiazepine withdrawal syndrome, which resembles symptoms during alcohol and barbiturate withdrawal. The higher the dose and the longer the drug is taken, the greater the risk of experiencing unpleasant withdrawal symptoms. Withdrawal symptoms can also occur from standard dosages and after short-term use. Abrupt withdrawal from therapeutic doses of temazepam after long-term use may result in a severe benzodiazepine withdrawal syndrome. Gradual and careful reduction of the dosage, preferably with a long-acting benzodiazepine with long half-life active metabolites, such as chlordiazepoxide or diazepam, are recommended to prevent severe withdrawal syndromes from developing. Other hypnotic benzodiazepines are not recommended. A study in rats found temazepam is cross tolerant with barbiturates and is able to effectively substitute for barbiturates and suppress barbiturate withdrawal signs. Rare cases are reported in the medical literature of psychotic states developing after abrupt withdrawal from benzodiazepines, even from therapeutic doses. Antipsychotics increase the severity of benzodiazepine withdrawal effects with an increase in the intensity and severity of convulsions. Patients who were treated in the hospital with temazepam or nitrazepam have continued taking these after leaving the hospital. Hypnotic uses in the hospital were recommended to be limited to five nights’ use only, to avoid the development of withdrawal symptoms such as insomnia.
Interactions
As with other benzodiazepines, temazepam produces additive CNS-depressant effects when co-administered with other medications which themselves produce CNS depression, such as barbiturates, alcohol, opiates, tricyclic antidepressants, nonselective MAO inhibitors, phenothiazines and other antipsychotics, skeletal muscle relaxants, antihistamines, and anaesthetics. Administration of theophylline or aminophylline has been shown to reduce the sedative effects of temazepam and other benzodiazepines.
Unlike many benzodiazepines, pharmacokinetic interactions involving the P450 system have not been observed with temazepam. Temazepam shows no significant interaction with CYP3A4 inhibitors (e.g. itraconazole, erythromycin). Oral contraceptives may decrease the effectiveness of temazepam and speed up its elimination half-life.
Overdose
Refer to Benzodiazepine Overdose.
Overdose (or an excess dose(s)) of temazepam results in increasing CNS effects, including:
- Somnolence (difficulty staying awake).
- Mental confusion.
- Respiratory depression.
- Hypotension.
- Impaired motor functions.
- Impaired or absent reflexes.
- Impaired coordination.
- Impaired balance.
- Dizziness, sedation.
- Coma.
- Death.
Temazepam had the highest rate of drug intoxication, including overdose, among common benzodiazepines in cases with and without combination with alcohol in a 1985 study. Temazepam and nitrazepam were the two benzodiazepines most commonly detected in overdose-related deaths in an Australian study of drug deaths. A 1993 British study found temazepam to have the highest number of deaths per million prescriptions among medications commonly prescribed in the 1980s (11.9, versus 5.9 for benzodiazepines overall, taken with or without alcohol).
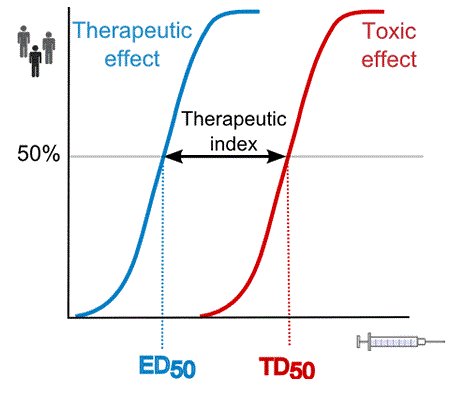
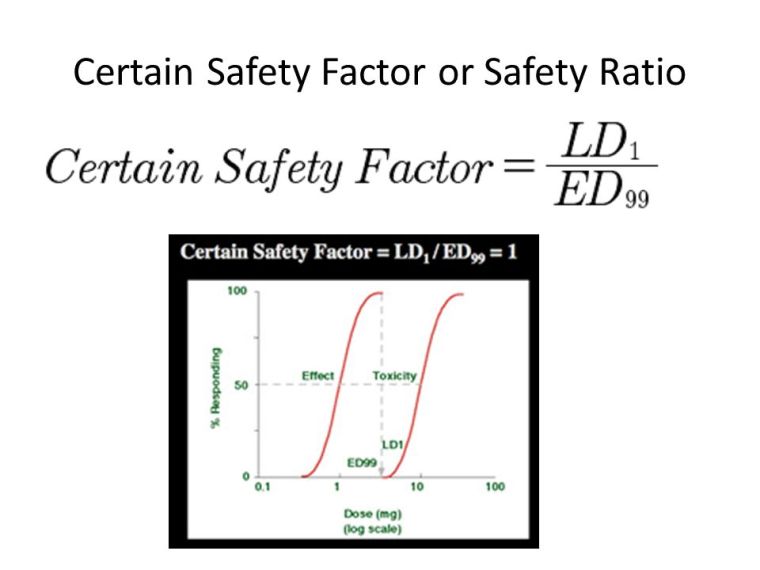
A 1995 Australian study of patients admitted to hospital after benzodiazepine overdose corroborated these results, and found temazepam overdose much more likely to lead to coma than other benzodiazepines (odds ratio 1.86). The authors noted several factors, such as differences in potency, receptor affinity, and rate of absorption between benzodiazepines, could explain this higher toxicity. Although benzodiazepines have a high therapeutic index, temazepam is one of the more dangerous of this class of drugs. The combination of alcohol and temazepam makes death by alcohol poisoning more likely.
Pharmacology
Temazepam is a white, crystalline substance, very slightly soluble in water, and sparingly soluble in alcohol. Its main pharmacological action is to increase the effect of the neurotransmitter gamma-aminobutyric acid (GABA) at the GABAA receptor. This causes sedation, motor impairment, ataxia, anxiolysis, an anticonvulsant effect, muscle relaxation, and a reinforcing effect. As a medication before surgery, temazepam decreased cortisol in elderly patients. In rats, it triggered the release of vasopressin into paraventricular nucleus of the hypothalamus and decreased the release of ACTH under stress.
Pharmacokinetics
Oral administration of 15 to 45 mg of temazepam in humans resulted in rapid absorption with significant blood levels achieved in fewer than 30 minutes and peak levels at two to three hours. In a single- and multiple-dose absorption, distribution, metabolism, and excretion (ADME) study, using tritium-labelled drug, temazepam was well absorbed and found to have minimal (8%) first-pass drug metabolism. No active metabolites were formed and the only significant metabolite present in blood was the O-conjugate. The unchanged drug was 96% bound to plasma proteins. The blood-level decline of the parent drug was biphasic, with the short half-life ranging from 0.4-0.6 hours and the terminal half-life from 3.5-18.4 hours (mean 8.8 hours), depending on the study population and method of determination.
Temazepam has very good bioavailability, with almost 100% being absorbed following being taken by mouth. The drug is metabolized through conjugation and demethylation prior to excretion. Most of the drug is excreted in the urine, with about 20% appearing in the faeces. The major metabolite was the O-conjugate of temazepam (90%); the O-conjugate of N-desmethyl temazepam was a minor metabolite (7%).
Society and Culture
Recreational Use
Refer to Benzodiazepine Use Disorder.
Temazepam is a drug with a moderate potential for misuse.
Benzodiazepines have been abused orally and intravenously. Different benzodiazepines have different abuse potential; the more rapid the increase in the plasma level following ingestion, the greater the intoxicating effect and the more open to abuse the drug becomes. The speed of onset of action of a particular benzodiazepine correlates well with the ‘popularity’ of that drug for abuse. The two most common reasons for preference were that a benzodiazepine was ‘strong’ and that it gave a good ‘high’.
A 1995 study found that temazepam is more rapidly absorbed and oxazepam is more slowly absorbed than most other benzodiazepines.
A 1985 study found that temazepam and triazolam maintained significantly higher rates of self-injection than a variety of other benzodiazepines. The study tested and compared the abuse liability of temazepam, triazolam, diazepam, lorazepam, oxazepam, flurazepam, alprazolam, chlordiazepoxide, clonazepam, nitrazepam, flunitrazepam, bromazepam, and clorazepate. The study tested self-injection rates on human, baboon, and rat subjects. All test subjects consistently showed a strong preference for temazepam and triazolam over all the rest of the benzodiazepines included in the study.
North America
In North America, temazepam misuse is not widespread. Other benzodiazepines are more commonly prescribed for insomnia. In the United States, temazepam is the fifth-most prescribed benzodiazepine, however there is a major drop off from the top four most prescribed (alprazolam, lorazepam, diazepam, and clonazepam in that order). Individuals abusing benzodiazepines obtain the drug by getting prescriptions from several doctors, forging prescriptions, or buying diverted pharmaceutical products on the illicit market. North America has never had a serious problem with temazepam misuse, but is becoming increasingly vulnerable to the illicit trade of temazepam.
Australia
Temazepam is a Schedule 4 drug and requires a prescription. The drug accounts for most benzodiazepine sought by forgery of prescriptions and through pharmacy burglary in Victoria. Due to rife intravenous abuse, the Australian government decided to put it under a more restrictive schedule than it had been, and since March 2004 temazepam capsules have been withdrawn from the Australian market, leaving only 10 mg tablets available. Benzodiazepines are commonly detected by Customs at different ports and airports, arriving by mail, also found occasionally in the baggage of air passengers, mostly small or medium quantities (up to 200-300 tablets) for personal use. From 2003 to 2006, customs detected about 500 illegal importations of benzodiazepines per year, most frequently diazepam. Quantities varied from single tablets to 2,000 tablets.
United Kingdom
In 1987, temazepam was the most widely abused legal prescription drug in the United Kingdom. The use of benzodiazepines by street-drug abusers was part of a polydrug abuse pattern, but many of those entering treatment facilities were declaring temazepam as their main drug of abuse. Temazepam was the most commonly used benzodiazepine in a study, published 1994, of injecting drug users in seven cities, and had been injected from preparations of capsules, tablets, and syrup. The increase in use of heroin, often mixed with other drugs, which most often included temazepam, diazepam, and alcohol, was a major factor in the increase in drug-related deaths in Glasgow and Edinburgh in 1990-1992. Temazepam use was particularly associated with violent or disorderly behaviours and contact with the police in a 1997 study of young single homeless people in Scotland. The BBC series Panorama featured an episode titled “Temazepam Wars”, dealing with the epidemic of temazepam abuse and directly related crime in Paisley, Scotland. The trend was mocked in the 1995 Black Grape song “Temazi Party” (also called “Tramazi Party”).
Medical Research Issues
The Journal of Clinical Sleep Medicine published a paper expressing concerns about benzodiazepine receptor agonist drugs, the benzodiazepines and the Z-drugs used as hypnotics in humans. The paper cites a systematic review of the medical literature concerning insomnia medications and states almost all trials of sleep disorders and drugs are sponsored by the pharmaceutical industry, while this is not the case in general medicine or psychiatry. It cites another study that “found that the odds ratio for finding results favourable to industry in industry-sponsored trials was 3.6 times as high as in non–industry-sponsored studies”. Issues discussed regarding industry-sponsored studies include: comparison of a drug to a placebo, but not to an alternative treatment; unpublished studies with unfavourable outcomes; and trials organized around a placebo baseline followed by drug treatment, but not counterbalanced with parallel-placebo-controlled studies. Quoting a 1979 report that too little research into hypnotics was independent of the drug manufacturers, the authors conclude, “the public desperately needs an equipoised assessment of hypnotic benefits and risks” and the NIH and VA should provide leadership to that end.
Street Terms
Street terms for temazepam include king kong pills (formerly referred to barbiturates, now more commonly refers to temazepam), jellies, jelly, Edinburgh eccies, tams, terms, mazzies, temazies, tammies, temmies, beans, eggs, green eggs, wobbly eggs, knockouts, hardball, norries, oranges (common term in Australia and New Zealand), rugby balls, ruggers, terminators, red and blue, no-gos, num nums, blackout, green devils, drunk pills, brainwash, mind erasers, neurotrashers, tem-tem’s (combined with buprenorphine), mommy’s big helper, vitamin T, big T, TZ, The Mazepam, Resties (North America) and others.
Availability
Temazepam is available in English-speaking countries under the following brand names:
- Euhypnos.
- Normison.
- Norkotral.
- Nortem.
- Remestan.
- Restoril.
- Temaze.
- Temtabs.
- Tenox.
In Spain, the drug is sold as ‘temzpem’. In Hungary the drug is sold as Signopam.
Legal Status
- In Austria, temazepam is listed in UN71 Schedule III under the Psychotropic Substances Decree of 1997.
- The drug is considered to have a high potential for abuse and addiction, but has accepted medical use for the treatment of severe insomnia.
- In Australia, temazepam is a Schedule 4 – Prescription Only medicine.
- It is primarily used for the treatment of insomnia, and is also seen as pre-anaesthetic medication.
- In Canada, temazepam is a Schedule IV controlled substance requiring a registered doctor’s prescription.
- In Denmark, temazepam is listed as a Class D substance under the Executive Order 698 of 1993 on Euphoric Substances which means it has a high potential for abuse, but is used for medical and scientific purposes.
- In Finland, temazepam is more tightly controlled than other benzodiazepines.
- The temazepam product Normison was pulled out of shelves and banned because the liquid inside gelatin capsules had caused a large increase in intravenous temazepam use.
- The other temazepam product, Tenox, was not affected and remains as prescription medicine.
- Temazepam intravenous use has not decreased to the level before Normison came to the market.
- In France, temazepam is listed as a psychotropic substance as are other similar drugs.
- It is prescribed with a non-renewable prescription (a new doctor visit every time), available only in 7 or 14-pill packaging for one or two weeks.
- One brand was withdrawn from the market in 2013.
- In Hong Kong, temazepam is regulated under Schedule 1 of Hong Kong’s Chapter 134 Dangerous Drugs Ordinance.
- Temazepam can only be used legally by health professionals and for university research purposes.
- The substance can be given by pharmacists under a prescription.
- Anyone who supplies the substance without prescription can be fined HKD$10,000.
- The penalty for trafficking or manufacturing the substance is a $5,000,000-fine and life imprisonment.
- Possession of the substance for consumption without license from the Department of Health is illegal with a $1,000,000-fine and/or seven years of jail time.
- In Ireland, temazepam is a Schedule 3 controlled substance with strict restrictions.
- In the Netherlands, temazepam is available for prescription as 10- or 20-mg tablets and capsules.
- Formulations of temazepam containing less than 20 mg are included in List 2 of the Opium Law, while formulations containing 20 mg or more of the drug (along with the gel-capsules) are a List 1 substance of the Opium Law, thus subject to more stringent regulation.
- Besides being used for insomnia, it is also occasionally used as a preanesthetic medication.
- In Norway, temazepam is not available as a prescription drug.
- It is regulated as a Class A substance under Norway’s Narcotics Act.
- In Portugal, temazepam is a Schedule IV controlled drug under Decree-Law 15/93.
- In Singapore, temazepam is a Class A controlled drug (Schedule I), making it illegal to possess and requiring a private prescription from a licensed physician to be dispensed.
- In Slovenia, it is regulated as a Group II (Schedule 2) controlled substance under the Production and Trade in Illicit Drugs Act.
- In South Africa, temazepam is a Schedule 5 drug, requiring a special prescription, and is restricted to 10- to 30-mg doses.
- In Sweden, temazepam is classed as a “narcotic” drug listed as both a List II (Schedule II) which denotes it is a drug with limited medicinal use and a high risk of addiction, and is also listed as a List V (Schedule V) substance which denotes the drug is prohibited in Sweden under the Narcotics Drugs Act (1968).
- Temazepam is banned in Sweden and possession and distribution of even small amounts is punishable by a prison sentence and a fine.
- In Switzerland, temazepam is a Class B controlled substance, like all other benzodiazepines.
- This means it is a prescription-only drug.
- In Thailand, temazepam is a Schedule II controlled drug under the Psychotropic Substances Act.
- Possession and distribution of the drug is illegal.
- In the United Kingdom, temazepam is a Class C controlled drug under the Misuse of Drugs Act 1971 (Schedule 3 under the Misuse of Drugs Regulations 2001).
- If prescribed privately (not on the NHS), temazepam is available only by a special controlled drug prescription form (FP10PCD) and pharmacies are obligated to follow special procedures for storage and dispensing.
- Additionally, all manufacturers in the UK have replaced the gel-capsules with solid tablets.
- Temazepam requires safe custody and up until June 2015 was exempt from CD prescription requirements.
- In the United States, Temazepam is currently a Schedule IV drug under the international Convention on Psychotropic Substances of 1971 and is only available by prescription.
- Specially coded prescriptions may be required in certain states.


You must be logged in to post a comment.