Introduction
According to a range of US governmental agencies devoted to healthcare studies, addiction and mental health disorders are deeply intertwined. It is not uncommon for someone seeking treatment for substance use to also be managing symptoms of depression, anxiety, trauma, or another psychiatric condition.

This combination is referred to as a co-occurring disorder or dual diagnosis. In Arizona specifically, the latest research from the Arizona Department of Human Services relays the following: 71 % of Arizona treatment providers reported offering dual‑diagnosis/co‑occurring services.
Understanding how common these conditions are, and how they interact, is key to getting the right help. Whether you are researching for a loved one or trying to make sense of your own experience, we provide a brief outline for you in this article.
We will review what you need to know about the prevalence, causes, and treatment of addiction alongside mental health disorders.
According to the Substance Abuse and Mental Health Services Administration, genetics significantly influence both mental illness and substance use disorders (SUDs). Shared genetic factors – such as those affecting brain reward systems – can increase risk for both conditions.
The Overlap Between Addiction and Mental Illness
Addiction does not occur in a vacuum. Many individuals who struggle with drugs or alcohol also experience underlying mental health conditions.
According to the National Institute on Drug Abuse “About half of the people who experience a substance use disorder also experience a mental illness at some point during their lifetime, and vice versa.”
In Arizona, this rate tends to be even higher. According to SAMHSA’s 2019 Behavioural Health Barometer, Arizona reports that 4-5% of adults experienced both SUD and any mental illness. These percentages surpass the national average of 3.8%.

Why Do These Conditions Co-Occur?
There are several reasons why mental health disorders and addiction commonly appear together:
- Self-medication: SAMHSA explains that mental health problems can lead some individuals to misuse substances “as a form of self‑medication” to alleviate distressing symptoms like anxiety or depression.
- Shared risk factors: Genetics contribute significantly, according to the National Centre for Biotechnology Information: Epigenetic changes triggered by trauma or stress can modify gene expression in ways that increase sensitivity to both mental health issues and substance use.
- Addiction-induced symptoms: SAMHSA states that substances “can cause people with an addiction to experience one or more symptoms of a mental health problem.” These symptoms may mirror anxiety, depression, psychosis, or mood disturbances during intoxication or withdrawal—and may persist until diagnosed and treated appropriately.
The relationship is rarely one-directional. Sometimes addiction leads to worsening mental health. Other times, unresolved trauma or an undiagnosed condition paves the way for substance use.
Common Mental Health Conditions Seen with Addiction
While co-occurring disorders can take many forms, certain psychiatric conditions are more frequently associated with substance use disorders.
Depression and Substance Use
Depression is among the most common co-occurring disorders. Nationally, SAMHSA states that depression is one of the most frequent mental–substance use co-occurring disorders, underscoring how individuals may self-medicate depressive symptoms with alcohol or sedatives, which then exacerbate depression over time.
Anxiety Disorders
Generalised anxiety disorder, panic disorder, and social phobia frequently appear alongside alcohol use, benzodiazepine misuse, or stimulant addiction. These substances can seem like a quick escape from anxiety but often reinforce the cycle of fear and dependence.
The National Institute of Mental Health confirms that GAD and social anxiety disorder are “commonly associated with alcohol and substance misuse”.
Post-Traumatic Stress Disorder (PTSD)
PTSD is closely linked to addiction, especially among veterans, first responders, and survivors of abuse. Arizona has a large population of military personnel and veterans who may face both PTSD and addiction, requiring trauma-informed, dual diagnosis care.
The US Air Force Medical Service materials state: withdrawal and stress can trigger PTSD symptoms (like flashbacks or hyperarousal), reinforcing substance use through emotional conditioning.
Bipolar Disorder
SAMHSA emphasizes that co-occurring SUD and bipolar symptoms create clinical ambiguity, often obscuring whether substance use or mood fluctuations came first.
Substance use can make it harder to diagnose and treat bipolar disorder accurately. During manic episodes, individuals may take risks with drugs or alcohol.
During depressive episodes, they may self-isolate or engage in harmful use patterns.
Schizophrenia and Psychotic Disorders
Though less common, individuals with schizophrenia or schizoaffective disorder can also struggle with substance use. According to the NCBI, NIDA and SAMHSA note that schizophrenia and other psychotic disorders, including schizoaffective disorder, are “highly prevalent” comorbid conditions with SUDs.
Specialized care is essential, particularly when symptoms of psychosis overlap with those caused by drug use (such as methamphetamine-induced psychosis, which has become more common in parts of Arizona). According to the Department of Justice, Methamphetamine abuse is increasing in Arizona, making more persons at risk for methamphetamine-induced psychosis.
Recognising the Signs of a Co-Occurring Disorder
It can be challenging to identify a co-occurring disorder—especially because addiction can mimic or mask psychiatric symptoms. Here are some warning signs SAMHSA indicates that a dual diagnosis might be present:
- Sudden mood swings or emotional numbness.
- Isolation from family and friends.
- Risky behaviour that escalates over time.
- Trouble managing daily responsibilities.
- Using substances to sleep, relax, or feel normal.
- History of trauma or prior psychiatric diagnosis.
In many cases, individuals with co-occurring disorders will not fully respond to addiction treatment alone unless their mental health needs are also addressed.
The Importance of Integrated Treatment in Arizona
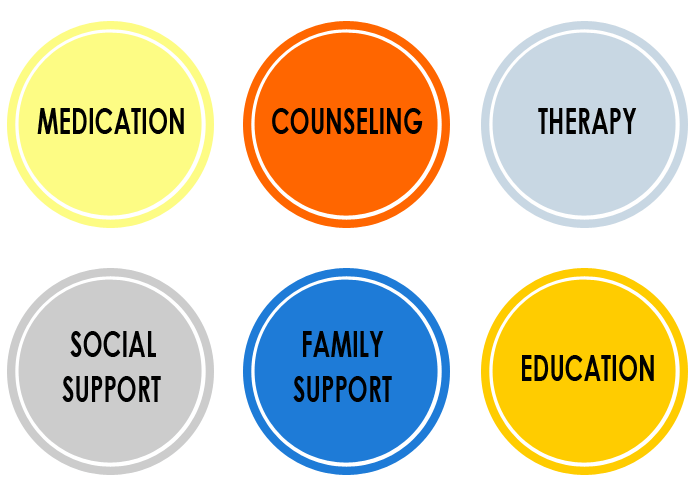
When both mental health and substance use disorders are present, integrated treatment is essential. This means treating both conditions at the same time, in the same setting, by the same clinical team.
Why Integrated Treatment Works
Research and clinical experience consistently show that individuals with co-occurring disorders do better when they receive:
- A comprehensive psychiatric evaluation.
- Medication management (when appropriate).
- Individual and group therapy focused on dual diagnosis.
- Psychoeducation about the interaction between mental health and addiction.
- Trauma-informed care and relapse prevention strategies.
In Arizona, dual diagnosis treatment is offered by specialized providers who understand the unique cultural and logistical barriers residents may face—especially those in rural areas or on AHCCCS (Arizona’s Medicaid programme). For support using AHCCCS, those struggling can find an accredited facility that handles trauma and addiction treatment in Phoenix.
Access to Care in Arizona
Arizona has expanded mental health and substance use services through various public and private efforts, including:
- AHCCCS coverage for dual diagnosis treatment at both inpatient and outpatient levels.
- Designated behavioural health facilities offering psychiatric stabilization and addiction care under one roof.
- Outreach efforts in underserved communities and tribal regions.
Still, waitlists and transportation issues remain barriers for some individuals, making early intervention all the more important.
Addressing Stigma Around Dual Diagnosis
Stigma remains one of the biggest obstacles to care. Some people may feel ashamed to seek help for either addiction or mental health concerns—let alone both at once. Families may misunderstand the symptoms and assume their loved one just needs “more willpower.”
The truth is that co-occurring disorders are medical conditions, not moral failings. Treatment works, and recovery is possible. In fact, when both mental health and addiction are addressed together, individuals are more likely to achieve long-term stability and improved quality of life.
What to Look for in a Dual Diagnosis Programme
If you or someone you care about in Arizona is dealing with both addiction and mental health challenges, finding the right treatment setting is key.
Look for programmes that offer:
- Medical detox with psychiatric support.
- A licensed mental health team (psychiatrists, therapists, counsellors).
- Evidence-based therapies like CBT, DBT, and EMDR.
- Support groups focused on co-occurring disorders.
- A structured discharge and aftercare plan.
Ask whether the programme accepts your insurance, especially if you are using AHCCCS, Health Choice, or another Arizona-based plan.
When to Seek Help
You don’t need to have everything “figured out” to start. Many people begin treatment unsure of whether they have a co-occurring diagnosis – and that is okay. A quality provider will help you uncover the full picture through assessment and ongoing care.
If substance use is interfering with your ability to function, and you have noticed symptoms of anxiety, depression, trauma, or mood instability, it is time to reach out. Waiting for things to get worse only increases the risk of crisis or overdose.
Summary
No matter where you are in the process: searching for answers, feeling stuck, or finally ready to act – help is available. With the right support, healing from both addiction and mental health struggles is not only possible but deeply rewarding.
If you are exploring options for dual diagnosis care in Arizona, do not hesitate to ask questions. A conversation with the right provider can open the door to lasting change: for you or your loved one.




You must be logged in to post a comment.