The Centre for Addiction and Mental Health (CAMH, French: Centre de toxicomanie et de santé mentale) is a psychiatric teaching hospital located in Toronto and ten community locations throughout the province of Ontario, Canada.
The hospital was formed in 1998 from the amalgamation of four separate institutions – the Queen Street Mental Health Centre, the Clarke Institute of Psychiatry, the Addiction Research Foundation, and the Donwood Institute. It is Canada’s largest mental health teaching hospital, and the only stand-alone psychiatric emergency department in Ontario. CAMH has 90 distinct clinical services across inpatient, outpatient, day treatment, and partial hospitalisation models. CAMH has been the site of major advancements in psychiatric research, including the discovery of the Dopamine receptor D2.
Brief History
CAMH was formed from the 1998 merger of the Queen Street Mental Health Centre with the Clarke Institute of Psychiatry, Addiction Research Foundation, and Donwood Institute.
Queen Street Mental Health Centre
The Provincial Lunatic Asylum opened on 26 January 1850. It was subsequently renamed Asylum for the Insane, then Hospital for the Insane, then Ontario Hospital (1919), and then the Queen Street Mental Health Centre (1966). It had also been called the Toronto Lunatic Asylum and 999 Queen Street West.
The original buildings were constructed in a series of rigid lines and sharp angles, consistent with the belief at the time that orderly physical structure would facilitate orderly mental states for the patients. High walls segregated the patients from the community, establishing a long-standing stigma about the facility.
The Queen Street Site of CAMH contained the Samuel A. Malcolmson Lecture Theatre, named for the site’s Chief Psychiatrist and Clinical Director of Forensics. In 2009, however, Malcolmson was subject to a disciplinary hearing of Ontario’s College of Physicians and Surgeons, following that he sexually abused a patient and fathered a child with her. Malcomson pleaded “no contest” and resigned his license to practice. The lecture hall was renamed the Queen Street Auditorium.
Reforms were made after a series of deaths at the Queen Street Mental Health Centre and newspaper accounts of involuntary drug treatment, electroshock therapy, and prison-like conditions.
Clarke Institute of Psychiatry
The institute was founded in 1966 and officially opened by Ontario Premier John P. Robarts. It was named the Clarke Institute of Psychiatry, after Charles Kirk Clarke, a pioneer in mental health in Canada. The institute took over the clinical, teaching, and research functions of the Toronto Psychiatric Hospital, located at 2 Surrey Place, which opened in 1925 under Clarence B. Farrar. The Institute served as the main psychiatry teaching hospital for the University of Toronto and was the headquarters for the Department of Psychiatry in the Faculty of Medicine. The first Medical and Executive Director of the Clarke was Charles Roberts, and the first Psychiatrist-in-Chief (and Professor and Head of the Department of Psychiatry at the University of Toronto) was Aldwyn B. Stokes. When Stokes retired in 1967, the administration was reorganised, and Robin Hunter became Medical Director and Psychiatrist-in-Chief.
Fredrick H. Lowy served as Psychiatrist-in-Chief of the Clarke from 1974 until 1980, followed by Vivian Rakoff from 1980 to 1990, and Paul E. Garfinkel from 1990 until the Clarke’s 1998 merger into CAMH.
Upon its merger into CAMH in 1998, the Clarke building become known as the CAMH College Street site. Conrad Black has been a generous supporter of the Clarke Institute of Psychiatry.
Rent Increase
In 2015, CAMH’s facilities at the College St. site were put in jeopardy following notice of a rent increase from $1.2 million to $4 million per year, at the renewal of its 20-year lease. CAMH valued its property at $25 million, whereas the hospital’s landlord, Brookfield Asset Management, valued it at $100 million. Brookfield turned down CBC’s interview requests. A government arbitrator was appointed who valued the property at $55 million, yielding a rent increase that CAMH was reportedly able to pay.
Addiction Research Foundation
The Addiction Research Foundation (ARF), then named the Alcoholism Research Foundation was founded in 1949, when H. David Archibald, who had studied at the School of Alcohol Studies at Yale University, was hired by the Liquor Control Board of Ontario. His mandate was to determine the scope of alcoholism in Ontario. He was named executive director when ARF opened and remained in that post until 1976. Focusing initially on outpatient treatment, their first facility was Brookside Hospital in 1951, expanding to branch offices and new locations in 1954, the same year they set up in-house research. In 1961, formally renamed the Alcoholism and Drug Addiction Research Foundation of Ontario, ARF expanded its mission to include drugs. In 1971, they expanded to a clinical teaching hospital called the Clinical Research and Treatment Institute. In 1978 ARF opened the School for Addiction Studies and expanded their international role in policy development and research. Following provincial hospital restructuring in the 1990s, ARF was folded in 1998 into CAMH.
Donwood Institute
In 1946, R. Gordon Bell opened a clinic in his own home for people needing mental health services. There existed an Ontario statute allowing doctors to take up to four patients into their homes without a hospital license. To his surprise, his only patients all suffered from alcoholism. It soon became necessary to move to larger facilities. To expand capacity, Bell opened Shadow Brook, which operated from 1947 to 1954, and then the Bell Clinic, which operated from 1954 to 1966. The Donwood Institute then opened in 1967, with 47 beds and a 4-month waiting list in the 1980s. Still focused on substance use, it claimed a 65% recovery rate for general population and an 85% recovery rate among physicians with addictions.
CAMH Queen St. Site Redevelopment
CAMH has been undergoing a three phase redevelopment centred at its Queen Street site, with four goals:
Deliver a new model of care and provide a healthy environment that promotes recovery;
Bring together the best research, clinical, education, health promotion, and policy experts in one place to change the future of mental health and addictions;
Revitalise the City of Toronto by opening up their site and by creating an inclusive new nine-block neighbourhood that benefits all; and
Change attitudes by breaking down barriers to eliminate the stigma of mental health.
Phase 1A was undertaken by Eastern Construction and completed in 2008 and Phase 1B was undertaken by Carillion and completed in 2012. Phase 1C is being undertaken by PCL Construction and is expected to be complete in 2020.
In 2008, CAMH announced the completion of four new buildings (forming Phase 1A of the project) to accommodate CAMH’s Addiction and Mood & Anxiety Programmes.
The Campbell Family Mental Health Research Institute was established with a $30 million donation in 2011 from Linda Campbell, Gaye Farncombe, and Susan Grange, each granddaughters of Canadian magnate Roy Thomson and nieces of Ken Thomson. CAMH CEO, Catherine Zahn, said research on the brain is the most promising pathway to progress in mental illness research. Bell donated $10 million to CAMH in 2011, reportedly the largest corporate gift in Canada to mental illness.
In 2012, CAMH announced the completion of three new buildings (forming Phase 1B of the project): the Bell Gateway Building for the central administration, a utilities and parking building, and the Intergenerational Wellness Centre which includes 12 new beds for youth ages 14–18.[28]
Margaret McCain, the former lieutenant-governor of New Brunswick and widow of McCain Foods co-founder Wallace McCain, donated $10 million to CAMH in 2012 to establish the Margaret and Wallace McCain Centre for Child, Youth and Family Mental Health. In 2013, the Slaight Family donated to $50 million to health care institutions, of which CAMH received $10 million. The donation permitted the opening of a centre dedicated to identifying and treating early signs of mental illness in youth.
In 2014, philanthropist and business executive Andrew Fass donated $1,000,000 to CAMH for the hospital to create a wellness program for its staff. In 2016, Fass withdrew his donation, saying that CAMH was unable to show how they were spending the money. The grant was to be used for CAMH’s “Well@Work” program, an initiative to provide Canadian workplaces with training to identify risks of mental illness and strategies to support employees in need.
CAMH opened the Emergency Department in 2014, funded by a $2.5 million private donation and a $4.2 million grant from the Ontario Ministry of Health and Long-Term Care.
In 2016, CAMH constructed a sweat lodge for Aboriginal patients in order to promote spiritual, physical, and emotional healing. Also in 2016, CAMH opened walk-in clinics for youth.
In 2020, the eight-storey, 110 patient bed McCain Complex Care & Recovery Building and five-storey 125 patient bed Crisis & Critical Care Building and 24-hour psychiatric emergency department were opened.
Research
CAMH reports being the largest research facility in Canada for mental health and addictions, including over 100 scientists over 150 research trainees. In the 2014-2015 fiscal year, CAMH received $44,384,230 in research funding and published more than 500 research articles.
Administration
Psychiatrist and Clarke Institute President Paul E. Garfinkel was appointed the first President and CEO of CAMH in 1998. He was followed by neurologist Catherine Zahn in 2009.[40]
Upon CAMH’s formation, Peter Catford was appointed vice president for Information Technology. In 2002, Catford outsourced the public hospital’s computer needs to H.I. Next, a private company which Catford founded and co-owned. When the Toronto Star reported on what it deemed an apparent conflict of interest regarding the spending of public money, the hospital would not reveal how much it paid Catford or his company, nor would CAMH disclose any details of its contract with H.I. Next or what other firms bid on the work. Catford commented only that “I feel honoured to work with (CAMH) and I feel like it has been done ethically.” In interviews with the Toronto Star, Dev Chopra, executive vice-president of CAMH first said there was nothing inappropriate about Catford’s role. “We got into it with our eyes open. There is no conflict.” However, Chopra later said there were “some optics from a conflict perspective” noting the hospital might revisit the issue that day. Catford left his CAMH position two days later, but the Star reported that hospital officials said changes were being considered months before the Star published its story about the issue.
Criticisms
CAMH’s administration has come under criticism including from staff who report safety problems and from donors who withdrew their funding citing accountability problems.
David Healy Affair
Soon after CAMH was founded, its administration was embroiled in a scandal involving Eli Lilly and Company, which donated $1.5 million to CAMH, and David Healy, a prominent critic of Prozac, the widely used antidepressant manufactured by Eli Lilly. CAMH hired Healy to be the head of its Mood and Anxiety Program, but withdrew the job offer after hearing about Healy’s views.
CAMH aggressively recruited Healy, and CAMH Physician-in-Chief, David Goldbloom, offered Healy a job as the head of the Mood and Anxiety Programme. Healy accepted and soon after gave a lecture in which he reiterated his views about Prozac increasing risk of suicide. A few days later, Goldbloom withdrew the job offer, saying “Essentially, we believe that it is not a good fit between you and the role as leader of an academic program in mood and anxiety disorders at the centre and in relation to the university….We do not feel your approach is compatible with the goals for development of the academic and clinical resource that we have.”
The decision caused an “uproar” among Canadian academics, with the Canadian Association of University Teachers calling CAMH actions “an affront to academic freedom in Canada.” Scientists from 13 countries, including Nobel laureates Julius Axelrod and Arvid Carlsson, protested CAMH’s actions as did the Society for Academic Freedom and Scholarship (SAFS).
Healy sued CAMH and the University of Toronto, alleging breach of contract, defamation, and denial of academic freedom. The lawsuit sought damages of $9.4 million, including $2.6 million from CAMH CEO Paul Garfinkel, and $1.4 million from the U of T Dean of Medicine. The university distanced itself from CAMH: According to U of T President, Robert Birgeneau, “Everyone is trying to blame the university for something that happened at one of our hospitals.”
The lawsuit was settled with Healy receiving an appointment as visiting professor as the University of Toronto. The president of the Canadian Association of University Teachers, Vic Catano, said “We see the settlement as a complete vindication for Dr. Healy.”
Child Gender Identity Clinic
In 1975, psychiatrist Susan Bradley founded a clinic in CAMH to work with gender dysphoric children. Bradley collaborated for many years with psychologist Kenneth Zucker, and they established the clinic as the largest gender identity service in Canada and an international centre for research. In their studies, 80% of the children grow out of the behaviour. They therefore use different approaches with children than adolescents because, over time, children are more likely to identify with their birth sex.
Regarding adolescents, Zucker “will support a teenager or adult who wants to transition using hormones and surgeries.” Regarding children, however, Zucker says “We are trying to help a child feel more comfortable with the gender identity that matches their birth sex” and that they use a variety of techniques to “help a child think more flexibly” about their gender. According to The New York Times, Zucker does this by “encouraging same-sex friendships and activities like board games that move beyond strict gender roles.” He said a child could be asked to make a list of pros and cons about being different genders so that the child realises that “there are both good and not so good things about being a boy and being a girl.”
Activists have criticized Zucker’s approach, claiming that it amounts to a form of conversion therapy. In 2015, following complaints from activists, CAMH commissioned an external review of the clinic. The review was inconclusive, reporting it “cannot state that the clinic does not practice reparative approaches.” Upon the release of the report, CAMH announced that it was closing the clinic and that Zucker was no longer at CAMH. Activists celebrated the news, calling upcoming community consultations “a major step toward establishing a service that will support families, and hopefully receive government funding to do so.” A petition of over 500 sexuality and gender diversity experts decried it, calling CAMH’s decisions “politically motivated” and showing an “indifference to research and scholarship.” In 2018, CAMH paid Zucker $586,000 and issued a public apology as part of a settlement.
Forensics
CAMH’s forensic department and its leadership have been the subject of criticism from Ontario judges and the public for issues including public safety, patient civil rights violations, and turning away patients ordered to CAMH by judges. This culminated in Ontario Superior Court Justice Maureen Forestell who reportedly “tore a strip off of” CAMH for its actions. Dr. Graham Glancy, a forensic psychiatrist at the Maplehurst Correctional Complex accused CAMH of focusing too much on patients and treatments to enhance its international reputation, but at the expense of the less glamorous forensic patients. He added “Cities like Ottawa and London are better resourced but Toronto is terrible. It’s a disgrace, really.” Toronto defence counsel Chris Hynes also attributed the problems to the CAMH leadership, saying “CAMH front-line workers pay the price for decisions made by the centre’s privatized board of directors.”
AWOL Patients
One recurrent issue has been the number of violent or dangerous patients who escape custody from CAMH’s forensic wards. After filing a freedom of information act request with the police, the Toronto Star reported that CAMH had nearly as many AWOLs as all other Toronto hospitals combined (2,060 and 2,371 for the period 2004 to 2014).
One of the most widely reported incidents involved Thomas Brailsford, who was institutionalised at CAMH after beheading his mother and being deemed a “danger to himself and others.” Brailsford took off from a taxicab on his way to a medical appointment, representing his second escape in a year.
In an interview with Toronto columnist Jerry Agar, CAMH’s chief of forensic psychiatry, Sandy Simpson said “Clearly, we will be reviewing this carefully to look at how we assessed the risk in this case.” Kate Richards of CAMH media relations subsequently said “We have tightened aspects of these procedures and…We are confident that the changes implemented in this case have been effective.” Agar wrote that he has “been eager to have Simpson back on the radio show to discuss what measures have been put in place” but had not received any response. Five months later, CAMH told Agar “Dr. Simpson is not available for a follow-up interview.”
In 2019, the Ontario government ordered a review of CAMH patient passes and privileges after a series of patient escapes from the secure forensic psychiatry units. In one case, a not criminally responsible (NCR) patient who absconded the hospital later left the country by plane.
Workplace Safety
In 2007, following a series of attacks on staff by patients, the Ontario Ministry of Labour asked CAMH to develop a workplace violence and policy programme. In 2008, the Ministry of Labour laid nine workplace safety charges against CAMH in response to allegations by staff that they had been attacked by patients. CAMH was fined $70,000 in 2009 for two attacks against nursing staff in 2007 and 2008. In 2014, the Ontario Ministry of Labour laid charges against CAMH for failing to protect workers from workplace violence following an attack earlier that year.
In 2014, the Ontario Ministry of Labour laid more charges against CAMH for failing to implement procedures to protect staff from workplace violence following another attack that year. The prosecution asked the court to send “a clear message” that the CAMH situation was unacceptable. CAMH was found guilty and fined again. CAMH’s Chief of Nursing, Rani Srivastava, said that CAMH accepted the court’s decision adding that the violence had a “devastating impact” on “all of us at CAMH.”
Three months later, another nurse was punched, kicked in the face, and dragged into a locked utility room where she was repeatedly kicked in the head, suffering fractures and nerve damage. Vicki McKenna of the Ontario Nurses’ Association reported that CAMH has not been taking part in government committees to improve workplace safety and called for CAMH senior management to be held personally responsible for the continued workplace violence. Rani Srivastava said that the incident was “completely unexpected,” that CAMH is “saddened” and “shocked” by what happened, and that “any incident is one too many.”
Reporting Failures
Simpson has also been criticised by the Ontario Review Board for secluding a patient for two months but not informing the board, as required. Simpson claimed that the regulation “was unclear” about whether CAMH had a duty to report what it was doing. The board disagreed, ruling against CAMH.
Public Policy Statements
CAMH issues statements about local and provincial regulations on addictive behaviours.
Alcohol
CAMH policy opposes the privatisation of alcohol sales, citing evidence that the increase in availability increases alcohol-related harms and associated costs. They supported that the Liquor Control Board of Ontario should maintain its monopoly on alcohol sales. CAMH referred to “the plan to allow the sale of VQA wines at farmers’ markets across the province” as “cause for concern” because it would increase access to alcohol. Similarly, together with other health organisations, CAMH called for a provincial alcohol strategy, ahead of Ontario’s plan to permit the sale of beer in grocery stores.
Marijuana
In a 2014 policy document, CAMH expressed support for the legalisation of marijuana with strict control regulations. According to CAMH CEO, Catherine Zahn, “Only through legalization can we implement a public-health approach, treating cannabis use as a health issue and not one to be addressed through law enforcement and the court system. This is the approach we take with tobacco and alcohol. As with alcohol, a legal cannabis market can be regulated with controls that address risk factors associated with use”.
Gambling
CAMH has opposed the expansion of Toronto’s Woodbine Racetrack. In a policy statement, CAMH said increased availability of gambling results in increased harms and predicted that a large portion of any increased revenues from the racetrack would come from people with gambling problems.
Censorship (psychoanalysis) (Zensur) is the force identified by Sigmund Freud as operating to separate consciousness from the unconscious mind.
In Dreaming
In his 1899 The Interpretation of Dreams, Freud identified a force working to disguise the dream-thoughts so as to make them more acceptable to the dreamer. In his wartime lectures, he compared its operation to the contemporary newspapers, where blanks would reveal first-hand the work of the censor, but where allusions, circumlocutions, and other softening techniques also showed attempts to work round the censorship of thoughts in advance. He went on to characterise the motivating force, which he called “the self-observing agency as the ego-censor [Zensor], the conscience; it is this that exercises the dream-censorship [Zensur] during the night, from which the repressions of inadmissable wishful impulses proceed”.
Another tool used by the dream-censorship was regression to archaic symbolic forms of expression unfamiliar to the conscious mind. Where all such measures of censorship failed, however, the result could be the development of nightmares and insomnia.
Psychoanalytic Extensions
Freud found the same effects of disguise and omission taking place in the construction of neurotic symptoms, under the influence of the censorship, as in dreams. He would eventually assign the role of censor to the mental agency he would term the superego.
Criticism
Sartre questioned how the censorship could operate unless it was already aware of the contents of the unconscious, and thought the phenomena Freud described could be better understood in terms of bad faith.
Milton Hyland Erickson (05 December 1901 to 25 March 1980) was an American psychiatrist and psychologist specializing in medical hypnosis and family therapy.
He was founding president of the American Society for Clinical Hypnosis and a fellow of the American Psychiatric Association, the American Psychological Association, and the American Psychopathological Association. He is noted for his approach to the unconscious mind as creative and solution-generating. He is also noted for influencing brief therapy, strategic family therapy, family systems therapy, solution focused brief therapy, and neuro-linguistic programming.
The Body Attitudes Test (BAT) was developed by Probst et al. in 1995. It was designed for the assessment of multiple eating disorders in women.
The BAT measures an individual’s subjective body experience and attitudes towards one’s own body. It is a questionnaire composed of twenty items which yields four different factors that evaluate the internal view of the patient’s own body.
The BAT is used to evaluate self-reported outlooks women with eating disorders have pre-, during, and post-treatment. It has been proven to highlight the psychological changes experienced throughout the rehabilitation process and is a useful way to gauge adherence and success of treatment.
This test also has the ability to differentiated between clinical and non-clinical subjects and between anorexics and bulimics. Studies have shown that patients suffering from restrictive anorexia have lower BAT scores, whereas patients with bulimia nervosa score higher.
Brief History
Michel Probst and colleagues began creating the BAT in 1984 and eventually published this questionnaire in 1995, with the goal of finding a new tool to evaluate how women suffering from eating disorders view their own body. The BAT was originally written in Dutch and then translated to many languages. This test was administered widely to both patients and control subjects, including women already diagnosed with eating disorders, women participating in Weight Watchers, and healthy women with no eating disorder diagnosis. To ensure the validity of this test, Probst and colleagues compared the results of the BAT to other tools already in use to evaluate women with eating disorders. These other evaluations include the Body Shape Questionnaire (BSQ), the Eating Disorder Inventory (EDI), and the Eating Attitudes Test (EAT).
Test
The BAT is a self-reported questionnaire consisting of 20 questions. Patients are asked to score each statement 0-5, 0 meaning they do not relate to the statement at all, and 5 meaning the statement frequently describes their sentiment. The following are examples of questions asked in the assessment:
I feel displeased when comparing my body to others.
I do not recognise my body as my own.
My body is too wide.
I am pleased with my body shape.
I feel the need to lose weight.
I see my breasts as too big.
I feel the need to conceal my body in looser clothing.
I avoid my reflection because it upsets me.
I do not struggle with relaxing.
I feel like every aspect of my body is broad.
My body negatively weighs on me.
There is a dissonance between my body and I.
At times, I feel like my body is swollen.
I feel threatened by my physical appearance.
I take great pride in my body size.
I feel like I look pregnant.
I always feel very tense.
I tend to be jealous of other people’s looks.
Aspects of my physical appearance scare me.
I often scrutinise my own reflection.
The answers to these questions are then analysed and provide information regarding four factors that evaluate the patient’s subjective view on their body:
The Ben-Tovim Walker Body Attitudes Questionnaire (BAQ) is a 44 item self-report questionnaire divided into six subscales that measures a woman’s attitude towards their own body.
The BAQ is used in the assessment of eating disorders. It was devised by D.I. Ben-Tovim and M.K. Walker in 1991.
The BAQ was the first body attitudes scale to be translated into Portuguese. The validity of the Portuguese language version was proven in a test conducted on a cohort of Brazilian women who speak Portuguese as their native language. The test-retest reliability was 0.57 and 0.85 after a one-month interval. The test was conducted by Scagliusi et al.
Japanese Version
The BAQ was translated into Japanese and tested on 68 males and 139 females in Japan and 68 Japanese males living in Australia (Kagawa et al.) The scores were assessed against 72 Australian men using the English-language version as well as scores from previous female Australian participants. There was a significant difference between the Japanese and Australian groups (p,0.05). The BAQ was deemed adequate for use in both Japanese males and females.
Scientific studies have found that different brain areas show altered activity in people with major depressive disorder (MDD), and this has encouraged advocates of various theories that seek to identify a biochemical origin of the disease, as opposed to theories that emphasize psychological or situational causes.
Factors spanning these causative groups include nutritional deficiencies in magnesium, vitamin D, and tryptophan with situational origin but biological impact. Several theories concerning the biologically based cause of depression have been suggested over the years, including theories revolving around monoamine neurotransmitters, neuroplasticity, neurogenesis, inflammation and the circadian rhythm. Physical illnesses, including hypothyroidism and mitochondrial disease, can also trigger depressive symptoms.
Neural circuits implicated in depression include those involved in the generation and regulation of emotion, as well as in reward. Abnormalities are commonly found in the lateral prefrontal cortex whose putative function is generally considered to involve regulation of emotion. Regions involved in the generation of emotion and reward such as the amygdala, anterior cingulate cortex (ACC), orbitofrontal cortex (OFC), and striatum are frequently implicated as well. These regions are innervated by a monoaminergic nuclei, and tentative evidence suggests a potential role for abnormal monoaminergic activity.
Genetic Factors
Difficulty of Gene Studies
Historically, candidate gene studies have been a major focus of study. However, as the number of genes reduces the likelihood of choosing a correct candidate gene, Type I errors (false positives) are highly likely. Candidate genes studies frequently possess a number of flaws, including frequent genotyping errors and being statistically underpowered. These effects are compounded by the usual assessment of genes without regard for gene-gene interactions. These limitations are reflected in the fact that no candidate gene has reached genome-wide significance.
Gene Candidates
5-HTTLPR
The 5-HTTLPR, or serotonin transporter promoter gene’s short allele, has been associated with increased risk of depression; since the 1990s, however, results have been inconsistent. Other genes that have been linked to a gene-environment interaction include CRHR1, FKBP5 and BDNF, the first two of which are related to the stress reaction of the HPA axis, and the latter of which is involved in neurogenesis. Candidate gene analysis of 5-HTTLPR on depression was inconclusive on its effect, either alone or in combination with life stress.
A 2003 study proposed that a gene-environment interaction (GxE) may explain why life stress is a predictor for depressive episodes in some individuals, but not in others, depending on an allelic variation of the serotonin-transporter-linked promoter region (5-HTTLPR). This hypothesis was widely-discussed in both the scientific literature and popular media, where it was dubbed the “Orchid gene”, but has conclusively failed to replicate in much larger samples, and the observed effect sizes in earlier work are not consistent with the observed polygenicity of depression.
BDNF
BDNF polymorphisms have also been hypothesized to have a genetic influence, but early findings and research failed to replicate in larger samples, and the effect sizes found by earlier estimates are inconsistent with the observed polygenicity of depression.
SIRT1 and LHPP
A 2015 GWAS study in Han Chinese women positively identified two variants in intronic regions near SIRT1 and LHPP with a genome-wide significant association.
Norepinephrine Transporter Polymorphisms
Attempts to find a correlation between norepinephrine transporter polymorphisms and depression have yielded negative results.
One review identified multiple frequently studied candidate genes. The genes encoding for the 5-HTT and 5-HT2A receptor were inconsistently associated with depression and treatment response. Mixed results were found for brain-derived neurotrophic factor (BDNF) Val66Met polymorphisms. Polymorphisms in the tryptophan hydroxylase gene was found to be tentatively associated with suicidal behaviour. A meta analysis of 182 case controlled genetic studies published in 2008 found Apolipoprotein E verepsilon 2 to be protective, and GNB3 825T, MTHFR 677T, SLC6A4 44bp insertion or deletions, and SLC6A3 40 bpVNTR 9/10 genotype to confer risk.
Circadian Rhythm
Depression may be related to abnormalities in the circadian rhythm, or biological clock.
A well synchronised circadian rhythm is critical for maintaining optimal health. Adverse changes and alterations in the circadian rhythm have been associated various neurological disorders and mood disorders including depression.
Depression may be related to the same brain mechanisms that control the cycles of sleep and wakefulness.
Sleep
Sleep disturbance is the most prominent symptom in depressive patients. Studies about sleep electroencephalograms have shown characteristic changes in depression such as reductions in non-rapid eye movement sleep production, disruptions of sleep continuity and disinhibition of rapid eye movement (REM) sleep. Rapid eye movement (REM) sleep – the stage in which dreaming occurs – may be quick to arrive and intense in depressed people. REM sleep depends on decreased serotonin levels in the brain stem, and is impaired by compounds, such as antidepressants, that increase serotonergic tone in brain stem structures. Overall, the serotonergic system is least active during sleep and most active during wakefulness. Prolonged wakefulness due to sleep deprivation activates serotonergic neurons, leading to processes similar to the therapeutic effect of antidepressants, such as the selective serotonin reuptake inhibitors (SSRIs). Depressed individuals can exhibit a significant lift in mood after a night of sleep deprivation. SSRIs may directly depend on the increase of central serotonergic neurotransmission for their therapeutic effect, the same system that impacts cycles of sleep and wakefulness.
Light Therapy
Research on the effects of light therapy on seasonal affective disorder suggests that light deprivation is related to decreased activity in the serotonergic system and to abnormalities in the sleep cycle, particularly insomnia. Exposure to light also targets the serotonergic system, providing more support for the important role this system may play in depression. Sleep deprivation and light therapy both target the same brain neurotransmitter system and brain areas as antidepressant drugs, and are now used clinically to treat depression. Light therapy, sleep deprivation and sleep time displacement (sleep phase advance therapy) are being used in combination quickly to interrupt a deep depression in people who are hospitalised for MDD.
Increased and decreased sleep length appears to be a risk factor for depression. People with MDD sometimes show diurnal and seasonal variation of symptom severity, even in non-seasonal depression. Diurnal mood improvement was associated with activity of dorsal neural networks. Increased mean core temperature was also observed. One hypothesis proposed that depression was a result of a phase shift.
Daytime light exposure correlates with decreased serotonin transporter activity, which may underlie the seasonality of some depression.
Monoamines
Monoamines are neurotransmitters that include serotonin, dopamine, norepinephrine, and epinephrine.
Illustration of the major elements in a prototypical synapse. Synapses are gaps between nerve cells. These cells convert their electrical impulses into bursts of chemical relayers, called neurotransmitters, which travel across the synapses to receptors on adjacent cells, triggering electrical impulses to travel down the latter cells.
Monoamine Hypothesis of Depression
Many antidepressant drugs acutely increase synaptic levels of the monoamine neurotransmitter, serotonin, but they may also enhance the levels of norepinephrine and dopamine. The observation of this efficacy led to the monoamine hypothesis of depression, which postulates that the deficit of certain neurotransmitters is responsible for depression, and even that certain neurotransmitters are linked to specific symptoms. Normal serotonin levels have been linked to mood and behaviour regulation, sleep, and digestion; norepinephrine to the fight-or-flight response; and dopamine to movement, pleasure, and motivation. Some have also proposed the relationship between monoamines and phenotypes such as serotonin in sleep and suicide, norepinephrine in dysphoria, fatigue, apathy, cognitive dysfunction, and dopamine in loss of motivation and psychomotor symptoms.[31] The main limitation for the monoamine hypothesis of depression is the therapeutic lag between initiation of antidepressant treatment and perceived improvement of symptoms. One explanation for this therapeutic lag is that the initial increase in synaptic serotonin is only temporary, as firing of serotonergic neurons in the dorsal raphe adapt via the activity of 5-HT1A autoreceptors. The therapeutic effect of antidepressants is thought to arise from autoreceptor desensitization over a period of time, eventually elevating firing of serotonergic neurons.
Serotonin
Initial studies of serotonin in depression examined peripheral measures such as the serotonin metabolite 5-Hydroxyindoleacetic acid (5-HIAA) and platelet binding. The results were generally inconsistent, and may not generalise to the central nervous system. However evidence from receptor binding studies and pharmacological challenges provide some evidence for dysfunction of serotonin neurotransmission in depression. Serotonin may indirectly influence mood by altering emotional processing biases that are seen at both the cognitive/behavioural and neural level. Pharmacologically reducing serotonin synthesis, and pharmacologically enhancing synaptic serotonin can produce and attenuate negative affective biases, respectively. These emotional processing biases may explain the therapeutic gap.
Dopamine
While various abnormalities have been observed in dopaminergic systems, results have been inconsistent. People with MDD have an increased reward response to dextroamphetamine compared to controls, and it has been suggested that this results from hypersensitivity of dopaminergic pathways due to natural hypoactivity. While polymorphisms of the D4 and D3 receptor have been implicated in depression, associations have not been consistently replicated. Similar inconsistency has been found in post-mortem studies, but various dopamine receptor agonists show promise in treating MDD. There is some evidence that there is decreased nigrostriatal pathway activity in people with melancholic depression (psychomotor retardation). Further supporting the role of dopamine in depression is the consistent finding of decreased cerebrospinal fluid and jugular metabolites of dopamine, as well as post mortem findings of altered Dopamine receptor D3 and dopamine transporter expression. Studies in rodents have supported a potential mechanism involving stress-induced dysfunction of dopaminergic systems.
Monoamine receptors affect phospholipase C and adenylyl cyclase inside of the cell. Green arrows means stimulation and red arrows inhibition. Serotonin receptors are blue, norepinephrine orange, and dopamine yellow. Phospholipase C and adenylyl cyclase start a signalling cascade which turn on or off genes in the cell. Sufficient ATP from mitochondria is required for these downstream signalling events. The 5HT-3 receptor is associated with gastrointestinal adverse effects and has no relationship to the other monoamine receptors.
Catecholamines
A number of lines of evidence indicative of decreased adrenergic activity in depression have been reported. Findings include the decreased activity of tyrosine hydroxylase, decreased size of the locus coeruleus, increased alpha 2 adrenergic receptor density, and decreased alpha 1 adrenergic receptor density. Furthermore, norepinephrine transporter knockout in mice models increases their tolerance to stress, implicating norepinephrine in depression.
One method used to study the role of monoamines is monoamine depletion. Depletion of tryptophan (the precursor of serotonin), tyrosine and phenylalanine (precursors to dopamine) does result in decreased mood in those with a predisposition to depression, but not in persons lacking the predisposition. On the other hand, inhibition of dopamine and norepinephrine synthesis with alpha-methyl-para-tyrosine does not consistently result in decreased mood.
Monoamine Oxidase
An offshoot of the monoamine hypothesis suggests that monoamine oxidase A (MAO-A), an enzyme which metabolises monoamines, may be overly active in depressed people. This would, in turn, cause the lowered levels of monoamines. This hypothesis received support from a PET study, which found significantly elevated activity of MAO-A in the brain of some depressed people. In genetic studies, the alterations of MAO-A-related genes have not been consistently associated with depression. Contrary to the assumptions of the monoamine hypothesis, lowered but not heightened activity of MAO-A was associated with depressive symptoms in adolescents. This association was observed only in maltreated youth, indicating that both biological (MAO genes) and psychological (maltreatment) factors are important in the development of depressive disorders. In addition, some evidence indicates that disrupted information processing within neural networks, rather than changes in chemical balance, might underlie depression.
Limitations
Since the 1990s, research has uncovered multiple limitations of the monoamine hypothesis, and its inadequacy has been criticised within the psychiatric community. For one thing, serotonin system dysfunction cannot be the sole cause of depression. Not all patients treated with antidepressants show improvements despite the usually rapid increase in synaptic serotonin. If significant mood improvements do occur, this is often not for at least two to four weeks. One possible explanation for this lag is that the neurotransmitter activity enhancement is the result of auto receptor desensitization, which can take weeks. Intensive investigation has failed to find convincing evidence of a primary dysfunction of a specific monoamine system in people with MDD. The antidepressants that do not act through the monoamine system, such as tianeptine and opipramol, have been known for a long time. There have also been inconsistent findings with regard to levels of serum 5-HIAA, a metabolite of serotonin. Experiments with pharmacological agents that cause depletion of monoamines have shown that this depletion does not cause depression in healthy people. Another problem that presents is that drugs that deplete monoamines may actually have antidepressant properties. Further, some have argued that depression may be marked by a hyperserotonergic state. Already limited, the monoamine hypothesis has been further oversimplified when presented to the general public.
Receptor Binding
As of 2012, efforts to determine differences in neurotransmitter receptor expression or for function in the brains of people with MDD using positron emission tomography (PET) had shown inconsistent results. Using the PET imaging technology and reagents available as of 2012, it appeared that the D1 receptor may be under-expressed in the striatum of people with MDD. 5-HT1A receptor binding literature is inconsistent; however, it leans towards a general decrease in the mesiotemporal cortex. 5-HT2A receptor binding appears to be unregulated in people with MDD. Results from studies on 5-HTT binding are variable, but tend to indicate higher levels in people with MDD. Results with D2/D3 receptor binding studies are too inconsistent to draw any conclusions. Evidence supports increased MAO activity in people with MDD, and it may even be a trait marker (not changed by response to treatment). Muscarinic receptor binding appears to be increased in depression, and, given ligand binding dynamics, suggests increased cholinergic activity.
Four meta analyses on receptor binding in depression have been performed, two on serotonin transporter (5-HTT), one on 5-HT1A, and another on dopamine transporter (DAT). One meta analysis on 5-HTT reported that binding was reduced in the midbrain and amygdala, with the former correlating with greater age, and the latter correlating with depression severity. Another meta-analysis on 5-HTT including both post-mortem and in vivo receptor binding studies reported that while in vivo studies found reduced 5-HTT in the striatum, amygdala and midbrain, post mortem studies found no significant associations. 5-HT1A was found to be reduced in the anterior cingulate cortex, mesiotemporal lobe, insula, and hippocampus, but not in the amygdala or occipital lobe. The most commonly used 5-HT1A ligands are not displaced by endogenous serotonin, indicating that receptor density or affinity is reduced. Dopamine transporter binding is not changed in depression.
Emotional Processing and Neural Circuits
Emotional Bias
People with MDD show a number of biases in emotional processing, such as a tendency to rate happy faces more negatively, and a tendency to allocate more attentional resources to sad expressions. Depressed people also have impaired recognition of happy, angry, disgusted, fearful and surprised, but not sad faces. Functional neuroimaging has demonstrated hyperactivity of various brain regions in response to negative emotional stimuli, and hypoactivity in response to positive stimuli. One meta analysis reported that depressed subjects showed decreased activity in the left dorsolateral prefrontal cortex and increased activity in the amygdala in response to negative stimuli. Another meta analysis reported elevated hippocampus and thalamus activity in a subgroup of depressed subjects who were medication naïve, not elderly, and had no comorbidities. The therapeutic lag of antidepressants has been suggested to be a result of antidepressants modifying emotional processing leading to mood changes. This is supported by the observation that both acute and sub-chronic SSRI administration increases response to positive faces. Antidepressant treatment appears to reverse mood congruent biases in limbic, prefrontal, and fusiform areas. dlPFC response is enhanced and amygdala response is attenuated during processing of negative emotions, the former or which is thought to reflect increased top down regulation. The fusiform gyrus and other visual processing areas respond more strongly to positive stimuli with antidepressant treatment, which is thought to reflect the a positive processing bias. These effects do not appear to be unique to serotonergic or noradrenergic antidepressants, but also occur in other forms of treatment such as deep brain stimulation.
Neural Circuits
One meta analysis of functional neuroimaging in depression observed a pattern of abnormal neural activity hypothesized to reflect an emotional processing bias. Relative to controls, people with MDD showed hyperactivity of circuits in the salience network (SN), composed of the pulvinar nuclei, the insula, and the dorsal anterior cingulate cortex (dACC), as well as decreased activity in regulatory circuits composed of the striatum and dlPFC.
A neuroanatomical model called the limbic-cortical model has been proposed to explain early biological findings in depression. The model attempts to relate specific symptoms of depression to neurological abnormalities. Elevated resting amygdala activity was proposed to underlie rumination, as stimulation of the amygdala has been reported to be associated with the intrusive recall of negative memories. The ACC was divided into pregenual (pgACC) and subgenual regions (sgACC), with the former being electrophysiologically associated with fear, and the latter being metabolically implicated in sadness in healthy subjects. Hyperactivity of the lateral orbitofrontal and insular regions, along with abnormalities in lateral prefrontal regions was suggested to underlie maladaptive emotional responses, given the regions roles in reward learning. This model and another termed “the cortical striatal model”, which focused more on abnormalities in the cortico-basal ganglia-thalamo-cortical loop, have been supported by recent literature. Reduced striatal activity, elevated OFC activity, and elevated sgACC activity were all findings consistent with the proposed models. However, amygdala activity was reported to be decreased, contrary to the limbic-cortical model. Furthermore, only lateral prefrontal regions were modulated by treatment, indicating that prefrontal areas are state markers (i.e. dependent upon mood), while subcortical abnormalities are trait markers (i.e. reflect a susceptibility).
Reward
While depression severity as a whole is not correlated with a blunted neural response to reward, anhedonia is directly correlated to reduced activity in the reward system. The study of reward in depression is limited by heterogeneity in the definition and conceptualisations of reward and anhedonia. Anhedonia is broadly defined as a reduced ability to feel pleasure, but questionnaires and clinical assessments rarely distinguish between motivational “wanting” and consummatory “liking”. While a number of studies suggest that depressed subjects rate positive stimuli less positively and as less arousing, a number of studies fail to find a difference. Furthermore, response to natural rewards such as sucrose does not appear to be attenuated. General affective blunting may explain “anhedonic” symptoms in depression, as meta analysis of both positive and negative stimuli reveal reduced rating of intensity. As anhedonia is a prominent symptom of depression, direct comparison of depressed with healthy subjects reveals increased activation of the subgenual anterior cingulate cortex (sgACC), and reduced activation of the ventral striatum, and in particular the nucleus accumbens (NAcc) in response to positive stimuli. Although the finding of reduced NAcc activity during reward paradigms is fairly consistent, the NAcc is made up of a functionally diverse range of neurons, and reduced blood-oxygen-level dependent (BOLD) signal in this region could indicate a variety of things including reduced afferent activity or reduced inhibitory output. Nevertheless, these regions are important in reward processing, and dysfunction of them in depression is thought to underlie anhedonia. Residual anhedonia that is not well targeted by serotonergic antidepressants is hypothesized to result from inhibition of dopamine release by activation of 5-HT2C receptors in the striatum. The response to reward in the medial orbitofrontal cortex (OFC) is attenuated in depression, while lateral OFC response is enhanced to punishment. The lateral OFC shows sustained response to absence of reward or punishment, and it is thought to be necessary for modifying behaviour in response to changing contingencies. Hypersensitivity in the lOFC may lead to depression by producing a similar effect to learned helplessness in animals.
Elevated response in the sgACC is a consistent finding in neuroimaging studies using a number of paradigms including reward related tasks. Treatment is also associated with attenuated activity in the sgACC, and inhibition of neurons in the rodent homologue of the sgACC, the infralimbic cortex (IL), produces an antidepressant effect. Hyperactivity of the sgACC has been hypothesized to lead to depression via attenuating the somatic response to reward or positive stimuli. Contrary to studies of functional magnetic resonance imaging response in the sgACC during tasks, resting metabolism is reduced in the sgACC. However, this is only apparent when correcting for the prominent reduction in sgACC volume associated with depression; structural abnormalities are evident at a cellular level, as neuropathological studies report reduced sgACC cell markers. The model of depression proposed from these findings by Drevets et al. suggests that reduced sgACC activity results in enhanced sympathetic nervous system activity and blunted HPA axis feedback. Activity in the sgACC may also not be causal in depression, as the authors of one review that examined neuroimaging in depressed subjects during emotional regulation hypothesized that the pattern of elevated sgACC activity reflected increased need to modulate automatic emotional responses in depression. More extensive sgACC and general prefrontal recruitment during positive emotional processing was associated with blunted subcortical response to positive emotions, and subject anhedonia. This was interpreted by the authors to reflect a downregulation of positive emotions by the excessive recruitment of the prefrontal cortex.
Neuroanatomy
While a number of neuroimaging findings are consistently reported in people with major depressive disorder, the heterogeneity of depressed populations presents difficulties interpreting these findings. For example, averaging across populations may hide certain subgroup related findings; while reduced dlPFC activity is reported in depression, a subgroup may present with elevated dlPFC activity. Averaging may also yield statistically significant findings, such as reduced hippocampal volumes, that are actually present in a subgroup of subjects. Due to these issues and others, including the longitudinal consistency of depression, most neural models are likely inapplicable to all depression.
Structural Neuroimaging
Meta analyses performed using seed-based d mapping have reported grey matter reductions in a number of frontal regions. One meta analysis of early onset general depression reported grey matter reductions in the bilateral anterior cingulate cortex (ACC) and dorsomedial prefrontal cortex (dmPFC). One meta analysis on first episode depression observed distinct patterns of grey matter reductions in medication free, and combined populations; medication free depression was associated with reductions in the right dorsolateral prefrontal cortex, right amygdala, and right inferior temporal gyrus; analysis on a combination of medication free and medicated depression found reductions in the left insula, right supplementary motor area, and right middle temporal gyrus. Another review distinguishing medicated and medication free populations, albeit not restricted to people with their first episode of MDD, found reductions in the combined population in the bilateral superior, right middle, and left inferior frontal gyrus, along with the bilateral parahippocampus. Increases in thalamic and ACC grey matter was reported in the medication free and medicated populations respectively. A meta analysis performed using “activation likelihood estimate” reported reductions in the paracingulate cortex, dACC and amygdala.
GMV reductions in MDD and BD.
Using statistical parametric mapping, one meta analysis replicated previous findings of reduced grey matter in the ACC, medial prefrontal cortex, inferior frontal gyrus, hippocampus and thalamus; however reductions in the OFC and ventromedial prefrontal cortex grey matter were also reported.
Two studies on depression from the ENIGMA consortium have been published, one on cortical thickness, and the other on subcortical volume. Reduced cortical thickness was reported in the bilateral OFC, ACC, insula, middle temporal gyri, fusiform gyri, and posterior cingulate cortices, while surface area deficits were found in medial occipital, inferior parietal, orbitofrontal and precentral regions. Subcortical abnormalities, including reductions in hippocampus and amygdala volumes, which were especially pronounced in early onset depression.
Multiple meta analysis have been performed on studies assessing white matter integrity using fractional anisotropy (FA). Reduced FA has been reported in the corpus callosum (CC) in both first episode medication naïve, and general major depressive populations. The extent of CC reductions differs from study to study. People with MDD who have not taken antidepressants before have been reported to have reductions only in the body of the CC and only in the genu of the CC. On the other hand, general MDD samples have been reported to have reductions in the body of the CC, the body and genu of the CC, and only the genu of the CC. Reductions of FA have also been reported in the anterior limb of the internal capsule (ALIC) and superior longitudinal fasciculus.
Functional Neuroimaging
Studies of resting state activity have utilised a number of indicators of resting state activity, including regional homogeneity (ReHO), amplitude of low frequency fluctuations (ALFF), fractional amplitude of low frequency fluctuations (fALFF), arterial spin labelling (ASL), and positron emission tomography measures of regional cerebral blood flow or metabolism.
MDD is associated with reduced FA in the ALIC and genu/body of the CC.
Studies using ALFF and fALFF have reported elevations in ACC activity, with the former primarily reporting more ventral findings, and the latter more dorsal findings. A conjunction analysis of ALFF and CBF studies converged on the left insula, with previously untreated people having increased insula activity. Elevated caudate CBF was also reported A meta analysis combining multiple indicators of resting activity reported elevated anterior cingulate, striatal, and thalamic activity and reduced left insula, post-central gyrus and fusiform gyrus activity. An activation likelihood estimate (ALE) meta analysis of PET/SPECT resting state studies reported reduced activity in the left insula, pregenual and dorsal anterior cingulate cortex and elevated activity in the thalamus, caudate, anterior hippocampus and amygdala. Compared to the ALE meta analysis of PET/SPECT studies, a study using multi-kernel density analysis reported hyperactivity only in the pulvinar nuclei of the thalamus.
Brain Regions
Research on the brains of people with MDD usually shows disturbed patterns of interaction between multiple parts of the brain. Several areas of the brain are implicated in studies seeking to more fully understand the biology of depression:
Subgenual Cingulate
Studies have shown that Brodmann area 25, also known as subgenual cingulate, is metabolically overactive in treatment-resistant depression. This region is extremely rich in serotonin transporters and is considered as a governor for a vast network involving areas like hypothalamus and brain stem, which influences changes in appetite and sleep; the amygdala and insula, which affect the mood and anxiety; the hippocampus, which plays an important role in memory formation; and some parts of the frontal cortex responsible for self-esteem. Thus disturbances in this area or a smaller than normal size of this area contributes to depression. Deep brain stimulation has been targeted to this region in order to reduce its activity in people with treatment resistant depression.
Prefrontal Cortex
One review reported hypoactivity in the prefrontal cortex of those with depression compared to controls. The prefrontal cortex is involved in emotional processing and regulation, and dysfunction of this process may be involved in the aetiology of depression. One study on antidepressant treatment found an increase in PFC activity in response to administration of antidepressants. One meta analysis published in 2012 found that areas of the prefrontal cortex were hypoactive in response to negative stimuli in people with MDD. One study suggested that areas of the prefrontal cortex are part of a network of regions including dorsal and pregenual cingulate, bilateral middle frontal gyrus, insula and superior temporal gyrus that appear to be hypoactive in people with MDD. However the authors cautioned that the exclusion criteria, lack of consistency and small samples limit results.
Amygdala
The amygdala, a structure involved in emotional processing appears to be hyperactive in those with major depressive disorder. The amygdala in unmedicated depressed persons tended to be smaller than in those that were medicated, however aggregate data shows no difference between depressed and healthy persons. During emotional processing tasks right amygdala is more active than the left, however there is no differences during cognitive tasks, and at rest only the left amygdala appears to be more hyperactive. One study, however, found no difference in amygdala activity during emotional processing tasks.
Hippocampus
Atrophy of the hippocampus has been observed during depression, consistent with animal models of stress and neurogenesis.
Stress can cause depression and depression-like symptoms through monoaminergic changes in several key brain regions as well as suppression in hippocampal neurogenesis. This leads to alteration in emotion and cognition related brain regions as well as HPA axis dysfunction. Through the dysfunction, the effects of stress can be exacerbated including its effects on 5-HT. Furthermore, some of these effects are reversed by antidepressant action, which may act by increasing hippocampal neurogenesis. This leads to a restoration in HPA activity and stress reactivity, thus restoring the deleterious effects induced by stress on 5-HT.
The hypothalamic-pituitary-adrenal axis is a chain of endocrine structures that are activated during the body’s response to stressors of various sorts. The HPA axis involves three structure, the hypothalamus which release CRH that stimulates the pituitary gland to release ACTH which stimulates the adrenal glands to release cortisol. Cortisol has a negative feedback effect on the pituitary gland and hypothalamus. In people with MDD this often shows increased activation in depressed people, but the mechanism behind this is not yet known. Increased basal cortisol levels and abnormal response to dexamethasone challenges have been observed in people with MDD. Early life stress has been hypothesized as a potential cause of HPA dysfunction. HPA axis regulation may be examined through a dexamethasone suppression tests, which tests the feedback mechanisms. Non-suppression of dexamethasone is a common finding in depression, but is not consistent enough to be used as a diagnostic tool. HPA axis changes may be responsible for some of the changes such as decreased bone mineral density and increased weight found in people with MDD. One drug, ketoconazole, currently under development has shown promise in treating MDD.
Hippocampal Neurogenesis
Reduced hippocampal neurogenesis leads to a reduction in hippocampal volume. A genetically smaller hippocampus has been linked to a reduced ability to process psychological trauma and external stress, and subsequent predisposition to psychological illness. Depression without familial risk or childhood trauma has been linked to a normal hippocampal volume but localised dysfunction.
Animal Models
A number of animal models exist for depression, but they are limited in that depression involves primarily subjective emotional changes. However, some of these changes are reflected in physiology and behaviour, the latter of which is the target of many animal models. These models are generally assessed according to four facets of validity; the reflection of the core symptoms in the model; the predictive validity of the model; the validity of the model with regard to human characteristics of aetiology; and the biological plausibility.
Different models for inducing depressive behaviours have been utilised; neuroanatomical manipulations such as olfactory bulbectomy or circuit specific manipulations with optogenetics; genetic models such as 5-HT1A knockout or selectively bred animals; models involving environmental manipulation associated with depression in humans, including chronic mild stress, early life stress and learned helplessness. The validity of these models in producing depressive behaviours may be assessed with a number of behavioural tests. Anhedonia and motivational deficits may, for example, be assessed via examining an animal’s level of engagement with rewarding stimuli such as sucrose or intracranial self-stimulation. Anxious and irritable symptoms may be assessed with exploratory behaviour in the presence of a stressful or novelty environment, such as the open field test, novelty suppressed feeding, or the elevated plus-maze. Fatigue, psychomotor poverty, and agitation may be assessed with locomotor activity, grooming activity, and open field tests.
Animal models possess a number of limitations due to the nature of depression. Some core symptoms of depression, such as rumination, low self-esteem, guilt, and depressed mood cannot be assessed in animals as they require subjective reporting. From an evolutionary standpoint, the behaviour correlates of defeats of loss are thought to be an adaptive response to prevent further loss. Therefore, attempts to model depression that seeks to induce defeat or despair may actually reflect adaption and not disease. Furthermore, while depression and anxiety are frequently comorbid, dissociation of the two in animal models is difficult to achieve. Pharmacological assessment of validity is frequently disconnected from clinical pharmacotherapeutics in that most screening tests assess acute effects, while antidepressants normally take a few weeks to work in humans.
Neurocircuits
Regions involved in reward are common targets of manipulation in animal models of depression, including the nucleus accumbens (NAc), ventral tegmental area (VTA), ventral pallidum (VP), lateral habenula (LHb) and medial prefrontal cortex (mPFC). Tentative fMRI studies in humans demonstrate elevated LHb activity in depression. The lateral habenula projects to the RMTg to drive inhibition of dopamine neurons in the VTA during omission of reward. In animal models of depression, elevated activity has been reported in LHb neurons that project to the ventral tegmental area (ostensibly reducing dopamine release). The LHb also projects to aversion reactive mPFC neurons, which may provide an indirect mechanism for producing depressive behaviours. Learned helplessness induced potentiation of LHb synapses are reversed by antidepressant treatment, providing predictive validity. A number of inputs to the LHb have been implicated in producing depressive behaviours. Silencing GABAergic projections from the NAc to the LHb reduces conditioned place preference induced in social aggression, and activation of these terminals induces CPP. Ventral pallidum firing is also elevated by stress induced depression, an effect that is pharmacologically valid, and silencing of these neurons alleviates behavioural correlates of depression. Tentative in vivo evidence from people with MDD suggests abnormalities in dopamine signalling. This led to early studies investigating VTA activity and manipulations in animal models of depression. Massive destruction of VTA neurons enhances depressive behaviours, while VTA neurons reduce firing in response to chronic stress. However, more recent specific manipulations of the VTA produce varying results, with the specific animal model, duration of VTA manipulation, method of VTA manipulation, and subregion of VTA manipulation all potentially leading to differential outcomes. Stress and social defeat induced depressive symptoms, including anhedonia, are associated with potentiation of excitatory inputs to Dopamine D2 receptor-expressing medium spiny neurons (D2-MSNs) and depression of excitatory inputs to Dopamine D1 receptor-expressing medium spiny neurons (D1-MSNs). Optogenetic excitation of D1-MSNs alleviates depressive symptoms and is rewarding, while the same with D2-MSNs enhances depressive symptoms. Excitation of glutaminergic inputs from the ventral hippocampus reduces social interactions, and enhancing these projections produces susceptibility to stress-induced depression. Manipulations of different regions of the mPFC can produce and attenuate depressive behaviours. For example, inhibiting mPFC neurons specifically in the intralimbic cortex attenuates depressive behaviours. The conflicting findings associated with mPFC stimulation, when compared to the relatively specific findings in the infralimbic cortex, suggest that the prelimbic cortex and infralimbic cortex may mediate opposing effects. mPFC projections to the raphe nuclei are largely GABAergic and inhibit the firing of serotonergic neurons. Specific activation of these regions reduce immobility in the forced swim test but do not affect open field or forced swim behaviour. Inhibition of the raphe shifts the behavioural phenotype of uncontrolled stress to a phenotype closer to that of controlled stress.
Altered Neuroplasticity
Recent studies have called attention to the role of altered neuroplasticity in depression. A review found a convergence of three phenomena:
Chronic stress reduces synaptic and dendritic plasticity;
Depressed subjects show evidence of impaired neuroplasticity (e.g. shortening and reduced complexity of dendritic trees); and
Anti-depressant medications may enhance neuroplasticity at both a molecular and dendritic level.
The conclusion is that disrupted neuroplasticity is an underlying feature of depression, and is reversed by antidepressants.
Blood levels of BDNF in people with MDD increase significantly with antidepressant treatment and correlate with decrease in symptoms. Post mortem studies and rat models demonstrate decreased neuronal density in the prefrontal cortex thickness in people with MDD. Rat models demonstrate histological changes consistent with MRI findings in humans, however studies on neurogenesis in humans are limited. Antidepressants appear to reverse the changes in neurogenesis in both animal models and humans.
Inflammation
Various reviews have found that general inflammation may play a role in depression. One meta analysis of cytokines in people with MDD found increased levels of pro-inflammatory IL-6 and TNF-a levels relative to controls. The first theories came about when it was noticed that interferon therapy caused depression in a large number of people receiving it. Meta analysis on cytokine levels in people with MDD have demonstrated increased levels of IL-1, IL-6, C-reactive protein, but not IL-10. Increased numbers of T-Cells presenting activation markers, levels of neopterin, IFN gamma, sTNFR, and IL-2 receptors have been observed in depression. Various sources of inflammation in depressive illness have been hypothesized and include trauma, sleep problems, diet, smoking and obesity. Cytokines, by manipulating neurotransmitters, are involved in the generation of sickness behaviour, which shares some overlap with the symptoms of depression. Neurotransmitters hypothesized to be affected include dopamine and serotonin, which are common targets for antidepressant drugs. Induction of indolamine-2,3 dioxygenease by cytokines has been proposed as a mechanism by which immune dysfunction causes depression. One review found normalization of cytokine levels after successful treatment of depression. A meta analysis published in 2014 found the use of anti-inflammatory drugs such as NSAIDs and investigational cytokine inhibitors reduced depressive symptoms. Exercise can act as a stressor, decreasing the levels of IL-6 and TNF-a and increasing those of IL-10, an anti-inflammatory cytokine.
Inflammation is also intimately linked with metabolic processes in humans. For example, low levels of Vitamin D have been associated with greater risk for depression. The role of metabolic biomarkers in depression is an active research area. Recent work has explored the potential relationship between plasma sterols and depressive symptom severity.
Oxidative Stress
A marker of DNA oxidation, 8-Oxo-2′-deoxyguanosine, has been found to be increased in both the plasma and urine of people with MDD. This along with the finding of increased F2-isoprostanes levels found in blood, urine and cerebrospinal fluid indicate increased damage to lipids and DNA in people with MDD. Studies with 8-Oxo-2′ Deoxyguanosine varied by methods of measurement and type of depression, but F2-Isoprostane level was consistent across depression types. Authors suggested lifestyle factors, dysregulation of the HPA axis, immune system and autonomics nervous system as possible causes. Another meta-analysis found similar results with regards to oxidative damage products as well as decreased oxidative capacity. Oxidative DNA damage may play a role in MDD.
Mitochondrial Dysfunction:
Increased markers of oxidative stress relative to controls have been found in people with MDD. These markers include high levels of RNS and ROS which have been shown to influence chronic inflammation, damaging the electron transport chain and biochemical cascades in mitochondria. This lowers the activity of enzymes in the respiratory chain resulting in mitochondrial dysfunction. The brain is a highly energy-consuming and has little capacity to store glucose as glycogen and so depends greatly on mitochondria. Mitochondrial dysfunction has been linked to the dampened neuroplasticity observed in depressed brains.
Large-Scale Brain Network Theory
Instead of studying one brain region, studying large scale brain networks is another approach to understanding psychiatric and neurological disorders, supported by recent research that has shown that multiple brain regions are involved in these disorders. Understanding the disruptions in these networks may provide important insights into interventions for treating these disorders. Recent work suggests that at least three large-scale brain networks are important in psychopathology.
Central Executive Network
The central executive network is made up of fronto-parietal regions, including dorsolateral prefrontal cortex and lateral posterior parietal cortex. This network is involved in high level cognitive functions such as maintaining and using information in working memory, problem solving, and decision making. Deficiencies in this network are common in most major psychiatric and neurological disorders, including depression. Because this network is crucial for everyday life activities, those who are depressed can show impairment in basic activities like test taking and being decisive.
Default Mode Network
The default mode network includes hubs in the prefrontal cortex and posterior cingulate, with other prominent regions of the network in the medial temporal lobe and angular gyrus. The default mode network is usually active during mind-wandering and thinking about social situations. In contrast, during specific tasks probed in cognitive science (for example, simple attention tasks), the default network is often deactivated. Research has shown that regions in the default mode network (including medial prefrontal cortex and posterior cingulate) show greater activity when depressed participants ruminate (that is, when they engage in repetitive self-focused thinking) than when typical, healthy participants ruminate. People with MDD also show increased connectivity between the default mode network and the subgenual cingulate and the adjoining ventromedial prefrontal cortex in comparison to healthy individuals, individuals with dementia or with autism. Numerous studies suggest that the subgenual cingulate plays an important role in the dysfunction that characterizes major depression. The increased activation in the default mode network during rumination and the atypical connectivity between core default mode regions and the subgenual cingulate may underlie the tendency for depressed individual to get “stuck” in the negative, self-focused thoughts that often characterise depression. However, further research is needed to gain a precise understanding of how these network interactions map to specific symptoms of depression.
Salience Network
The salience network is a cingulate-frontal operculum network that includes core nodes in the anterior cingulate and anterior insula. A salience network is a large-scale brain network involved in detecting and orienting the most pertinent of the external stimuli and internal events being presented. Individuals who have a tendency to experience negative emotional states (scoring high on measures of neuroticism) show an increase in the right anterior insula during decision-making, even if the decision has already been made. This atypically high activity in the right anterior insula is thought to contribute to the experience of negative and worrisome feelings. In MDD, anxiety is often a part of the emotional state that characterises depression.
1897 – Wilhelm Reich, Austrian-American psychotherapist and academic (d. 1957).
Wilhelm Reich
Wilhelm Reich (24 March 1897 to 03 November 1957) was an Austrian doctor of medicine and a psychoanalyst, along with being a member of the second generation of analysts after Sigmund Freud. The author of several influential books, most notably The Impulsive Character (1925), Character Analysis (1933), and The Mass Psychology of Fascism (1933), he became known as one of the most radical figures in the history of psychiatry.
Reich’s work on character contributed to the development of Anna Freud’s The Ego and the Mechanisms of Defence (1936), and his idea of muscular armour – the expression of the personality in the way the body moves shaped innovations such as body psychotherapy, Gestalt therapy, bioenergetic analysis and primal therapy. His writing influenced generations of intellectuals; he coined the phrase “the sexual revolution” and according to one historian acted as its midwife. During the 1968 student uprisings in Paris and Berlin, students scrawled his name on walls and threw copies of The Mass Psychology of Fascism at police.
After graduating in medicine from the University of Vienna in 1922, Reich became deputy director of Freud’s outpatient clinic, the Vienna Ambulatorium. During the 1930s, he was part of a general trend among younger analysts and Frankfurt sociologists that tried to reconcile psychoanalysis with Marxism. He is credited for establishing the first sexual advisory clinics in Vienna, along with Marie Frischauf. He said he wanted to “attack the neurosis by its prevention rather than treatment”.
He moved to New York in 1939, after having accepted a position as Assistant Professor at the New School of Social Research. During his five years in Oslo, he had coined the term “orgone energy” – from “orgasm” and “organism” – for the notion of life energy. In 1940 he started building orgone accumulators, modified Faraday cages that he claimed were beneficial for cancer patients. He claimed that his laboratory cancer mice had had remarkable positive effects from being kept in a Faraday cage, so he built human-size versions, where one could sit inside. This led to newspaper stories about “sex boxes” that cured cancer.
Following two critical articles about him in The New Republic and Harper’s in 1947, the US Food and Drug Administration obtained an injunction against the interstate shipment of orgone accumulators and associated literature, believing they were dealing with a “fraud of the first magnitude”. Charged with contempt in 1956 for having violated the injunction, Reich was sentenced to two years imprisonment, and that summer over six tons of his publications were burned by order of the court. He died in prison of heart failure just over a year later, days before he was due to apply for parole.
Nancy Coover Andreasen (born 11 November 1938) is an American neuroscientist and neuropsychiatrist.
She currently holds the Andrew H. Woods Chair of Psychiatry at the Roy J. and Lucille A. Carver College of Medicine at the University of Iowa.
Early Life
Andreasen was born in Lincoln, Nebraska. She received her undergraduate degree from the University of Nebraska with majors in English, History, and Philosophy. She received a Ph.D. in English literature. She was a Professor of Renaissance Literature in the Department of English at the University of Iowa for 5 years. She published scholarly articles on John Donne and her first book in the field of Renaissance English literature: John Donne: Conservative Revolutionary
Clinical
A serious illness after the birth of her first daughter piqued Andreasen’s interest in medicine and biomedical research, and she decided to change careers to study medicine. She attended medical school at the University of Iowa College of Medicine, graduated in 1970 and completed her psychiatry residency in 1973. In 1974, she conducted the first modern empirical study of creativity that recognised some association between creativity and manic-depressive illness.
Early in her career she recognised that negative symptoms and associated cognitive impairments had more debilitating effects than psychotic symptoms, like delusions and hallucinations. While psychotic symptoms represent an exaggeration of normal brain/mind functions, negative symptoms represent a loss of normal functions, for example, alogia the loss of the ability to think and speak fluently, affective blunting the loss of the ability to express emotions, avolition, loss of the ability to initiate goal-directed activity, and anhedonia, loss of the ability to experience emotions. The papers describing these concepts have become citation classics, as determined by the Science Citation Index produced by the Institute for Scientific Information. Andreasen is largely responsible for development of the concept of negative symptoms in schizophrenia, having created the first widely used scales for rating the positive and negative symptoms of schizophrenia. She became one of the world’s foremost authorities on schizophrenia. She contributed to nosology and phenomenology by serving on the DSM III and DSM IV Task Forces, chairing the Schizophrenia Work Group for DSM IV.
Andreasen pioneered the application of neuroimaging techniques in major mental illnesses, and published the first quantitative study of magnetic resonance imaging (MRI) of brain abnormalities in schizophrenia. Andreasen became director of the Iowa Mental Health Clinical Research Centre and the Psychiatric Iowa Neuroimaging Consortium. She leads a multidisciplinary team working on three-dimensional image analysis techniques to integrate multi-modality imaging and on developing automated analysis of structural and functional imaging techniques. Software developed by this team is known as BRAINS (Brain Research: Analysis of Images, Networks, and Systems).
She resumed research about the neuroscience of creativity in the 2000s.
Honours
In 2000 President Clinton awarded her the National Medal of Science, America’s highest award for scientific achievement. This award was given for:
her pivotal contributions to the social and behavioral sciences, through the integrative study of mind, brain, and behavior, by joining behavioral science with the technologies of neuroscience and neuroimaging in order to understand mental processes such as memory and creativity, and mental illnesses such as schizophrenia.
She has received numerous other awards, including the Interbrew-Baillet-Latour Prize from the Belgian Academy of Science, the Lieber Schizophrenia Research Prize, and many awards from the American Psychiatric Association, including its Research Prize, the Judd Marmor Award, and the Distinguished Service Award. She was elected a Fellow of the American Academy of Arts and Sciences in 2002. She is a member of the National Academy of Medicine (formerly the Institute of Medicine of the National Academy of Sciences. She was elected to serve two terms on the governing council of the latter organisation. She chaired two Institute of Medicine/National Academy of Sciences Committees that published influential reports. She served as Editor-in-Chief of the American Journal of Psychiatry for 13 years. She is past president of the American Psychopathological Association and the Psychiatric Research Society. She was the founding Chair of the Neuroscience Section of the American Association for the Advancement of Science. She is a member of the Society for Neuroscience and on the Honorary International Editorial Advisory Board of the Mens Sana Monographs.
Experience of Sexism
She has spoken about her experiences of sexism. Early in her career she found that her articles were more likely to be accepted for publication when she used her initials instead of her first name.
Personal Life
She is the mother of two daughters. Suz Andreasen, who was a jewellry designer who lived in New York City, died from ovarian cancer on 10 November 2010. Robin Andreasen is a professor of Cognitive Science at the University of Delaware. She is married to Captain Terry Gwinn, a retired military officer who flew helicopter gunships for 3.5 tours during the Vietnam War.
Selected Bibliography
She has written three books for the general public:
“The Broken Brain: The Biological Revolution in Psychiatry” (1983).
“Brave New Brain: Conquering Mental Illness in the Era of the Genome” (2001).
“The Creating Brain: The Neuroscience of Genius”.
She authored, co-authored, or edited twelve other scholarly books and over 600 articles.
John Donne: Conservative Revolutionary. 1967.
Introductory Textbook of Psychiatry, Fourth Edition by Nancy C. Andreasen and Donald W. Black.
Understanding mental illness: A layman’s guide (Religion and medicine series).
Schizophrenia: From Mind to Molecule (American Psychopathological Association).
Within psychological testing, the Scale for the Assessment of Positive Symptoms (SAPS) is a rating scale to measure positive symptoms in schizophrenia.
The scale was developed by Nancy Andreasen and was first published in 1984. SAPS is split into 4 domains, and within each domain separate symptoms are rated from 0 (absent) to 5 (severe). The scale is closely linked to the Scale for the Assessment of Negative Symptoms (SANS) which was published a few years earlier.
Negative symptoms are those conspicuous by their absence – lack of concern for one’s appearance, and lack of language and communication skills, for example. Nancy Andreasen developed the scale and first published it in 1984. SANS splits assessment into five domains. Within each domain it rates separate symptoms from 0 (absent) to 5 (severe). The scale is closely linked to the Scale for the Assessment of Positive Symptoms (SAPS), which was published a few years later. These tools are available for clinicians and for research.
Background
Schizophrenia is a severe mental illness characterised by a range of behaviours, including hallucinations and delusions. Hallucinations refer to disorders involving the sensory systems, and are most often manifested as seeing or hearing things (e.g. voices) that do not exist. Delusions include odd or unusual beliefs such as grandiosity or paranoia. Both hallucinations and delusions are inconsistent with reality. Other symptoms of schizophrenia include bizarre behaviour, odd posture or movements, facial grimacing, loss of, or indifference to self-help skills (grooming, washing, toileting, feeding, etc.). Schizophrenia may also be marked by a host of social and communication deficits, such as social withdrawal, odd use of language, including excessive use of made up words (neologisms), incomprehensible combinations of words (word salad) or overall poverty of speech. The symptoms are often classified into two broad categories: positive and negative symptoms. Positive symptoms refer to those behaviours or condition that are present in schizophrenia but that are not present under typical conditions (hallucinations, delusions). Negative symptoms refer to those behaviours that are conspicuous because of their absence (grooming, language, communication). Several measures or rating scales have been developed to assess the positive and negative aspects of schizophrenia.



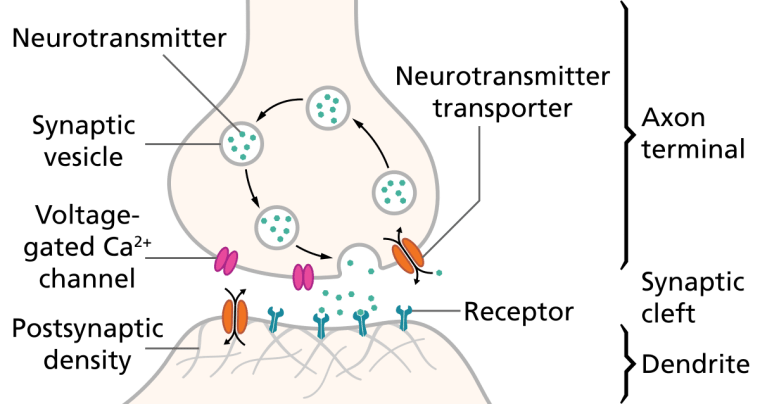
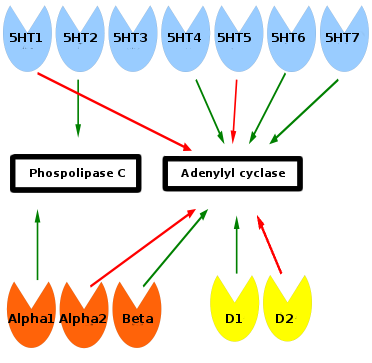


You must be logged in to post a comment.